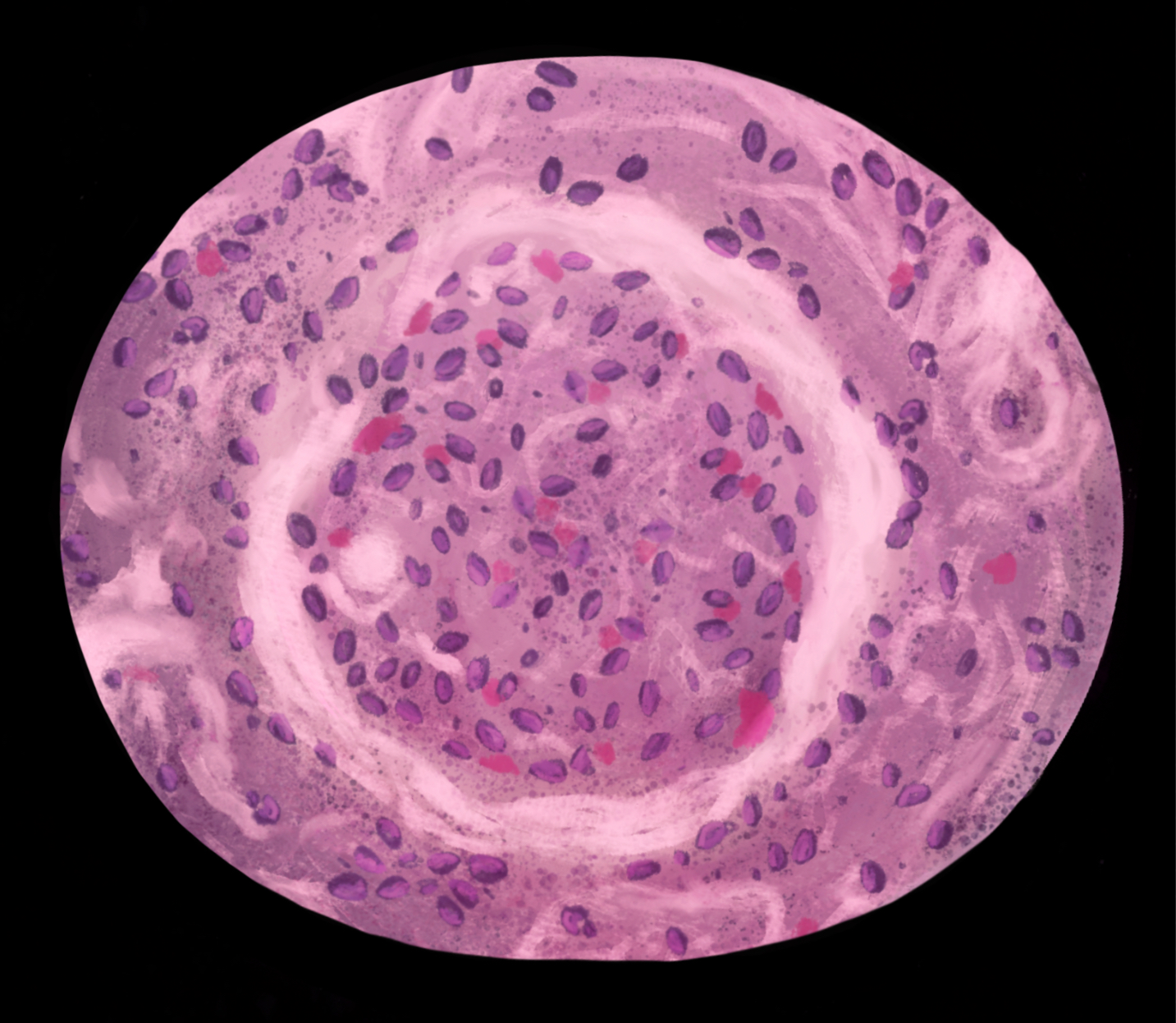
[1]
Pattrapornpisut P, Avila-Casado C, Reich HN. IgA Nephropathy: Core Curriculum 2021. American journal of kidney diseases : the official journal of the National Kidney Foundation. 2021 Sep:78(3):429-441. doi: 10.1053/j.ajkd.2021.01.024. Epub 2021 Jul 9
[PubMed PMID: 34247883]
[2]
Working Group of the International IgA Nephropathy Network and the Renal Pathology Society, Coppo R, Troyanov S, Camilla R, Hogg RJ, Cattran DC, Cook HT, Feehally J, Roberts IS, Amore A, Alpers CE, Barratt J, Berthoux F, Bonsib S, Bruijn JA, D'Agati V, D'Amico G, Emancipator SN, Emma F, Ferrario F, Fervenza FC, Florquin S, Fogo AB, Geddes CC, Groene HJ, Haas M, Herzenberg AM, Hill PA, Hsu SI, Jennette JC, Joh K, Julian BA, Kawamura T, Lai FM, Li LS, Li PK, Liu ZH, Mezzano S, Schena FP, Tomino Y, Walker PD, Wang H, Weening JJ, Yoshikawa N, Zhang H. The Oxford IgA nephropathy clinicopathological classification is valid for children as well as adults. Kidney international. 2010 May:77(10):921-7. doi: 10.1038/ki.2010.43. Epub 2010 Mar 3
[PubMed PMID: 20200498]
[3]
Shen PC, He LQ, Tang Y, Wang Q, Wang W, Li J. Clinicopathological characteristics and prognostic factors of asymptomatic IgA nephropathy. Journal of investigative medicine : the official publication of the American Federation for Clinical Research. 2010 Mar:58(3):560-5. doi: 10.231/JIM.0b013e3181d20aa1. Epub
[PubMed PMID: 20215916]
[4]
Rodrigues JC, Haas M, Reich HN. IgA Nephropathy. Clinical journal of the American Society of Nephrology : CJASN. 2017 Apr 3:12(4):677-686. doi: 10.2215/CJN.07420716. Epub 2017 Feb 3
[PubMed PMID: 28159829]
[5]
Rollino C, Vischini G, Coppo R. IgA nephropathy and infections. Journal of nephrology. 2016 Aug:29(4):463-8. doi: 10.1007/s40620-016-0265-x. Epub 2016 Jan 22
[PubMed PMID: 26800970]
[6]
Woo KT, Lau YK, Choong HL, Tan HK, Foo MW, Lee EJ, Anantharaman V, Lee GS, Yap HK, Yi Z, Fook-Chong S, Wong KS, Chan CM. Genomics and disease progression in IgA nephritis. Annals of the Academy of Medicine, Singapore. 2013 Dec:42(12):674-80
[PubMed PMID: 24463829]
[7]
Lai KN, Tang SC, Schena FP, Novak J, Tomino Y, Fogo AB, Glassock RJ. IgA nephropathy. Nature reviews. Disease primers. 2016 Feb 11:2():16001. doi: 10.1038/nrdp.2016.1. Epub 2016 Feb 11
[PubMed PMID: 27189177]
[8]
Han SH, Kang EW, Kie JH, Yoo TH, Choi KH, Han DS, Kang SW. Spontaneous remission of IgA nephropathy associated with resolution of hepatitis A. American journal of kidney diseases : the official journal of the National Kidney Foundation. 2010 Dec:56(6):1163-7. doi: 10.1053/j.ajkd.2010.08.018. Epub 2010 Oct 8
[PubMed PMID: 20932622]
[9]
Soares MF, Roberts IS. IgA nephropathy: an update. Current opinion in nephrology and hypertension. 2017 May:26(3):165-171. doi: 10.1097/MNH.0000000000000312. Epub
[PubMed PMID: 28221174]
Level 3 (low-level) evidence
[10]
McGrogan A, Franssen CF, de Vries CS. The incidence of primary glomerulonephritis worldwide: a systematic review of the literature. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association. 2011 Feb:26(2):414-30. doi: 10.1093/ndt/gfq665. Epub 2010 Nov 10
[PubMed PMID: 21068142]
Level 1 (high-level) evidence
[11]
Utsunomiya Y, Koda T, Kado T, Okada S, Hayashi A, Kanzaki S, Kasagi T, Hayashibara H, Okasora T. Incidence of pediatric IgA nephropathy. Pediatric nephrology (Berlin, Germany). 2003 Jun:18(6):511-5
[PubMed PMID: 12720079]
[12]
Schena FP, Nistor I. Epidemiology of IgA Nephropathy: A Global Perspective. Seminars in nephrology. 2018 Sep:38(5):435-442. doi: 10.1016/j.semnephrol.2018.05.013. Epub
[PubMed PMID: 30177015]
Level 3 (low-level) evidence
[13]
Kiryluk K, Li Y, Sanna-Cherchi S, Rohanizadegan M, Suzuki H, Eitner F, Snyder HJ, Choi M, Hou P, Scolari F, Izzi C, Gigante M, Gesualdo L, Savoldi S, Amoroso A, Cusi D, Zamboli P, Julian BA, Novak J, Wyatt RJ, Mucha K, Perola M, Kristiansson K, Viktorin A, Magnusson PK, Thorleifsson G, Thorsteinsdottir U, Stefansson K, Boland A, Metzger M, Thibaudin L, Wanner C, Jager KJ, Goto S, Maixnerova D, Karnib HH, Nagy J, Panzer U, Xie J, Chen N, Tesar V, Narita I, Berthoux F, Floege J, Stengel B, Zhang H, Lifton RP, Gharavi AG. Geographic differences in genetic susceptibility to IgA nephropathy: GWAS replication study and geospatial risk analysis. PLoS genetics. 2012:8(6):e1002765. doi: 10.1371/journal.pgen.1002765. Epub 2012 Jun 21
[PubMed PMID: 22737082]
[14]
Barbour SJ, Cattran DC, Kim SJ, Levin A, Wald R, Hladunewich MA, Reich HN. Individuals of Pacific Asian origin with IgA nephropathy have an increased risk of progression to end-stage renal disease. Kidney international. 2013 Nov:84(5):1017-24. doi: 10.1038/ki.2013.210. Epub 2013 Jun 5
[PubMed PMID: 23739233]
[15]
Magistroni R, D'Agati VD, Appel GB, Kiryluk K. New developments in the genetics, pathogenesis, and therapy of IgA nephropathy. Kidney international. 2015 Nov:88(5):974-89. doi: 10.1038/ki.2015.252. Epub 2015 Sep 16
[PubMed PMID: 26376134]
[16]
Suzuki H, Kiryluk K, Novak J, Moldoveanu Z, Herr AB, Renfrow MB, Wyatt RJ, Scolari F, Mestecky J, Gharavi AG, Julian BA. The pathophysiology of IgA nephropathy. Journal of the American Society of Nephrology : JASN. 2011 Oct:22(10):1795-803. doi: 10.1681/ASN.2011050464. Epub 2011 Sep 23
[PubMed PMID: 21949093]
[17]
Kiryluk K, Moldoveanu Z, Sanders JT, Eison TM, Suzuki H, Julian BA, Novak J, Gharavi AG, Wyatt RJ. Aberrant glycosylation of IgA1 is inherited in both pediatric IgA nephropathy and Henoch-Schönlein purpura nephritis. Kidney international. 2011 Jul:80(1):79-87. doi: 10.1038/ki.2011.16. Epub 2011 Feb 16
[PubMed PMID: 21326171]
[18]
Gharavi AG, Moldoveanu Z, Wyatt RJ, Barker CV, Woodford SY, Lifton RP, Mestecky J, Novak J, Julian BA. Aberrant IgA1 glycosylation is inherited in familial and sporadic IgA nephropathy. Journal of the American Society of Nephrology : JASN. 2008 May:19(5):1008-14. doi: 10.1681/ASN.2007091052. Epub 2008 Feb 13
[PubMed PMID: 18272841]
[19]
Knoppova B, Reily C, Maillard N, Rizk DV, Moldoveanu Z, Mestecky J, Raska M, Renfrow MB, Julian BA, Novak J. The Origin and Activities of IgA1-Containing Immune Complexes in IgA Nephropathy. Frontiers in immunology. 2016:7():117. doi: 10.3389/fimmu.2016.00117. Epub 2016 Apr 12
[PubMed PMID: 27148252]
[20]
Lamm ME, Emancipator SN, Robinson JK, Yamashita M, Fujioka H, Qiu J, Plaut AG. Microbial IgA protease removes IgA immune complexes from mouse glomeruli in vivo: potential therapy for IgA nephropathy. The American journal of pathology. 2008 Jan:172(1):31-6. doi: 10.2353/ajpath.2008.070131. Epub 2007 Dec 28
[PubMed PMID: 18165266]
Level 2 (mid-level) evidence
[21]
Huang ZQ, Raska M, Stewart TJ, Reily C, King RG, Crossman DK, Crowley MR, Hargett A, Zhang Z, Suzuki H, Hall S, Wyatt RJ, Julian BA, Renfrow MB, Gharavi AG, Novak J. Somatic Mutations Modulate Autoantibodies against Galactose-Deficient IgA1 in IgA Nephropathy. Journal of the American Society of Nephrology : JASN. 2016 Nov:27(11):3278-3284
[PubMed PMID: 26966014]
[22]
Yamamoto R, Nagasawa Y, Shoji T, Iwatani H, Hamano T, Kawada N, Inoue K, Uehata T, Kaneko T, Okada N, Moriyama T, Horio M, Yamauchi A, Tsubakihara Y, Imai E, Rakugi H, Isaka Y. Cigarette smoking and progression of IgA nephropathy. American journal of kidney diseases : the official journal of the National Kidney Foundation. 2010 Aug:56(2):313-24. doi: 10.1053/j.ajkd.2010.02.351. Epub 2010 May 14
[PubMed PMID: 20471735]
[23]
Kataoka H, Ohara M, Honda K, Mochizuki T, Nitta K. Maximal glomerular diameter as a 10-year prognostic indicator for IgA nephropathy. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association. 2011 Dec:26(12):3937-43. doi: 10.1093/ndt/gfr139. Epub 2011 Mar 22
[PubMed PMID: 21427079]
[24]
Zhai YL, Zhu L, Shi SF, Liu LJ, Lv JC, Zhang H. Increased APRIL Expression Induces IgA1 Aberrant Glycosylation in IgA Nephropathy. Medicine. 2016 Mar:95(11):e3099. doi: 10.1097/MD.0000000000003099. Epub
[PubMed PMID: 26986150]
[25]
Kiryluk K, Li Y, Scolari F, Sanna-Cherchi S, Choi M, Verbitsky M, Fasel D, Lata S, Prakash S, Shapiro S, Fischman C, Snyder HJ, Appel G, Izzi C, Viola BF, Dallera N, Del Vecchio L, Barlassina C, Salvi E, Bertinetto FE, Amoroso A, Savoldi S, Rocchietti M, Amore A, Peruzzi L, Coppo R, Salvadori M, Ravani P, Magistroni R, Ghiggeri GM, Caridi G, Bodria M, Lugani F, Allegri L, Delsante M, Maiorana M, Magnano A, Frasca G, Boer E, Boscutti G, Ponticelli C, Mignani R, Marcantoni C, Di Landro D, Santoro D, Pani A, Polci R, Feriozzi S, Chicca S, Galliani M, Gigante M, Gesualdo L, Zamboli P, Battaglia GG, Garozzo M, Maixnerová D, Tesar V, Eitner F, Rauen T, Floege J, Kovacs T, Nagy J, Mucha K, Pączek L, Zaniew M, Mizerska-Wasiak M, Roszkowska-Blaim M, Pawlaczyk K, Gale D, Barratt J, Thibaudin L, Berthoux F, Canaud G, Boland A, Metzger M, Panzer U, Suzuki H, Goto S, Narita I, Caliskan Y, Xie J, Hou P, Chen N, Zhang H, Wyatt RJ, Novak J, Julian BA, Feehally J, Stengel B, Cusi D, Lifton RP, Gharavi AG. Discovery of new risk loci for IgA nephropathy implicates genes involved in immunity against intestinal pathogens. Nature genetics. 2014 Nov:46(11):1187-96. doi: 10.1038/ng.3118. Epub 2014 Oct 12
[PubMed PMID: 25305756]
[26]
Roos A, Rastaldi MP, Calvaresi N, Oortwijn BD, Schlagwein N, van Gijlswijk-Janssen DJ, Stahl GL, Matsushita M, Fujita T, van Kooten C, Daha MR. Glomerular activation of the lectin pathway of complement in IgA nephropathy is associated with more severe renal disease. Journal of the American Society of Nephrology : JASN. 2006 Jun:17(6):1724-34
[PubMed PMID: 16687629]
[27]
Roos A, Bouwman LH, van Gijlswijk-Janssen DJ, Faber-Krol MC, Stahl GL, Daha MR. Human IgA activates the complement system via the mannan-binding lectin pathway. Journal of immunology (Baltimore, Md. : 1950). 2001 Sep 1:167(5):2861-8
[PubMed PMID: 11509633]
[28]
Maillard N, Wyatt RJ, Julian BA, Kiryluk K, Gharavi A, Fremeaux-Bacchi V, Novak J. Current Understanding of the Role of Complement in IgA Nephropathy. Journal of the American Society of Nephrology : JASN. 2015 Jul:26(7):1503-12. doi: 10.1681/ASN.2014101000. Epub 2015 Feb 18
[PubMed PMID: 25694468]
Level 3 (low-level) evidence
[29]
Gharavi AG, Kiryluk K, Choi M, Li Y, Hou P, Xie J, Sanna-Cherchi S, Men CJ, Julian BA, Wyatt RJ, Novak J, He JC, Wang H, Lv J, Zhu L, Wang W, Wang Z, Yasuno K, Gunel M, Mane S, Umlauf S, Tikhonova I, Beerman I, Savoldi S, Magistroni R, Ghiggeri GM, Bodria M, Lugani F, Ravani P, Ponticelli C, Allegri L, Boscutti G, Frasca G, Amore A, Peruzzi L, Coppo R, Izzi C, Viola BF, Prati E, Salvadori M, Mignani R, Gesualdo L, Bertinetto F, Mesiano P, Amoroso A, Scolari F, Chen N, Zhang H, Lifton RP. Genome-wide association study identifies susceptibility loci for IgA nephropathy. Nature genetics. 2011 Mar 13:43(4):321-7. doi: 10.1038/ng.787. Epub 2011 Mar 13
[PubMed PMID: 21399633]
[30]
Zhu L, Zhai YL, Wang FM, Hou P, Lv JC, Xu DM, Shi SF, Liu LJ, Yu F, Zhao MH, Novak J, Gharavi AG, Zhang H. Variants in Complement Factor H and Complement Factor H-Related Protein Genes, CFHR3 and CFHR1, Affect Complement Activation in IgA Nephropathy. Journal of the American Society of Nephrology : JASN. 2015 May:26(5):1195-204. doi: 10.1681/ASN.2014010096. Epub 2014 Sep 9
[PubMed PMID: 25205734]
[31]
Hassler JR. IgA nephropathy: A brief review. Seminars in diagnostic pathology. 2020 May:37(3):143-147. doi: 10.1053/j.semdp.2020.03.001. Epub 2020 Mar 16
[PubMed PMID: 32241578]
[32]
Kusaba G, Ohsawa I, Ishii M, Inoshita H, Takagi M, Tanifuji C, Takahashi K, Nakamoto J, Yoshida M, Ohi H, Horikoshi S, Kurihara H, Tomino Y. Significance of broad distribution of electron-dense deposits in patients with IgA nephropathy. Medical molecular morphology. 2012 Dec:45(1):29-34. doi: 10.1007/s00795-011-0538-3. Epub 2012 Mar 20
[PubMed PMID: 22431181]
[34]
Shen P, He L, Huang D. Clinical course and prognostic factors of clinical early IgA nephropathy. The Netherlands journal of medicine. 2008 Jun:66(6):242-7
[PubMed PMID: 18689907]
[35]
D'Amico G. Natural history of idiopathic IgA nephropathy and factors predictive of disease outcome. Seminars in nephrology. 2004 May:24(3):179-96
[PubMed PMID: 15156525]
[36]
Lai KN, Lai FM, Chan KW, Ho CP, Leung AC, Vallance-Owen J. An overlapping syndrome of IgA nephropathy and lipoid nephrosis. American journal of clinical pathology. 1986 Dec:86(6):716-23
[PubMed PMID: 3538845]
[37]
Herlitz LC, Bomback AS, Stokes MB, Radhakrishnan J, D'Agati VD, Markowitz GS. IgA nephropathy with minimal change disease. Clinical journal of the American Society of Nephrology : CJASN. 2014 Jun 6:9(6):1033-9. doi: 10.2215/CJN.11951113. Epub 2014 Apr 10
[PubMed PMID: 24721885]
[38]
Walsh M, Sar A, Lee D, Yilmaz S, Benediktsson H, Manns B, Hemmelgarn B. Histopathologic features aid in predicting risk for progression of IgA nephropathy. Clinical journal of the American Society of Nephrology : CJASN. 2010 Mar:5(3):425-30. doi: 10.2215/CJN.06530909. Epub 2010 Jan 14
[PubMed PMID: 20089495]
[39]
Pozzi C. Treatment of IgA nephropathy. Journal of nephrology. 2016 Feb:29(1):21-5. doi: 10.1007/s40620-015-0248-3. Epub 2015 Nov 17
[PubMed PMID: 26577268]
[40]
Gharavi AG, Yan Y, Scolari F, Schena FP, Frasca GM, Ghiggeri GM, Cooper K, Amoroso A, Viola BF, Battini G, Caridi G, Canova C, Farhi A, Subramanian V, Nelson-Williams C, Woodford S, Julian BA, Wyatt RJ, Lifton RP. IgA nephropathy, the most common cause of glomerulonephritis, is linked to 6q22-23. Nature genetics. 2000 Nov:26(3):354-7
[PubMed PMID: 11062479]
[41]
Lv J, Xu D, Perkovic V, Ma X, Johnson DW, Woodward M, Levin A, Zhang H, Wang H, TESTING Study Group. Corticosteroid therapy in IgA nephropathy. Journal of the American Society of Nephrology : JASN. 2012 Jun:23(6):1108-16. doi: 10.1681/ASN.2011111112. Epub 2012 Apr 26
[PubMed PMID: 22539830]
[42]
Coppo R, Treatment of IgA nephropathy: Recent advances and prospects. Nephrologie
[PubMed PMID: 29606258]
Level 3 (low-level) evidence
[43]
Lai KN, Leung JC, Tang SC. The Treatment of IgA Nephropathy. Kidney diseases (Basel, Switzerland). 2015 May:1(1):19-26. doi: 10.1159/000381508. Epub 2015 Apr 15
[PubMed PMID: 27536661]
[44]
Courtney AE, McNamee PT, Nelson WE, Maxwell AP. Does angiotensin blockade influence graft outcome in renal transplant recipients with IgA nephropathy? Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association. 2006 Dec:21(12):3550-4
[PubMed PMID: 16968729]
[45]
Rauen T, Eitner F, Fitzner C, Sommerer C, Zeier M, Otte B, Panzer U, Peters H, Benck U, Mertens PR, Kuhlmann U, Witzke O, Gross O, Vielhauer V, Mann JF, Hilgers RD, Floege J, STOP-IgAN Investigators. Intensive Supportive Care plus Immunosuppression in IgA Nephropathy. The New England journal of medicine. 2015 Dec 3:373(23):2225-36. doi: 10.1056/NEJMoa1415463. Epub
[PubMed PMID: 26630142]
[46]
Kittiskulnam P,Kanjanabuch T,Tangmanjitjaroen K,Chancharoenthana W,Praditpornsilpa K,Eiam-Ong S, The beneficial effects of weight reduction in overweight patients with chronic proteinuric immunoglobulin a nephropathy: a randomized controlled trial. Journal of renal nutrition : the official journal of the Council on Renal Nutrition of the National Kidney Foundation. 2014 May;
[PubMed PMID: 24759301]
Level 1 (high-level) evidence
[47]
Pozzi C, Bolasco PG, Fogazzi GB, Andrulli S, Altieri P, Ponticelli C, Locatelli F. Corticosteroids in IgA nephropathy: a randomised controlled trial. Lancet (London, England). 1999 Mar 13:353(9156):883-7
[PubMed PMID: 10093981]
Level 1 (high-level) evidence
[48]
Manno C, Torres DD, Rossini M, Pesce F, Schena FP. Randomized controlled clinical trial of corticosteroids plus ACE-inhibitors with long-term follow-up in proteinuric IgA nephropathy. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association. 2009 Dec:24(12):3694-701. doi: 10.1093/ndt/gfp356. Epub 2009 Jul 23
[PubMed PMID: 19628647]
Level 1 (high-level) evidence
[49]
Lafayette RA, Canetta PA, Rovin BH, Appel GB, Novak J, Nath KA, Sethi S, Tumlin JA, Mehta K, Hogan M, Erickson S, Julian BA, Leung N, Enders FT, Brown R, Knoppova B, Hall S, Fervenza FC. A Randomized, Controlled Trial of Rituximab in IgA Nephropathy with Proteinuria and Renal Dysfunction. Journal of the American Society of Nephrology : JASN. 2017 Apr:28(4):1306-1313. doi: 10.1681/ASN.2016060640. Epub 2016 Nov 7
[PubMed PMID: 27821627]
Level 1 (high-level) evidence
[50]
Beck L, Bomback AS, Choi MJ, Holzman LB, Langford C, Mariani LH, Somers MJ, Trachtman H, Waldman M. KDOQI US commentary on the 2012 KDIGO clinical practice guideline for glomerulonephritis. American journal of kidney diseases : the official journal of the National Kidney Foundation. 2013 Sep:62(3):403-41. doi: 10.1053/j.ajkd.2013.06.002. Epub 2013 Jul 18
[PubMed PMID: 23871408]
Level 2 (mid-level) evidence
[51]
Appel GB, Contreras G, Dooley MA, Ginzler EM, Isenberg D, Jayne D, Li LS, Mysler E, Sánchez-Guerrero J, Solomons N, Wofsy D, Aspreva Lupus Management Study Group. Mycophenolate mofetil versus cyclophosphamide for induction treatment of lupus nephritis. Journal of the American Society of Nephrology : JASN. 2009 May:20(5):1103-12. doi: 10.1681/ASN.2008101028. Epub 2009 Apr 15
[PubMed PMID: 19369404]
[52]
Chen Y, Li Y, Yang S, Li Y, Liang M. Efficacy and safety of mycophenolate mofetil treatment in IgA nephropathy: a systematic review. BMC nephrology. 2014 Dec 5:15():193. doi: 10.1186/1471-2369-15-193. Epub 2014 Dec 5
[PubMed PMID: 25475967]
Level 1 (high-level) evidence
[53]
Tian L, Shao X, Xie Y, Wang L, Wang Q, Che X, Ni Z, Mou S. The long-term efficacy and safety of immunosuppressive therapy on the progression of IgA nephropathy: a meta-analysis of controlled clinical trials with more than 5-year follow-up. Expert opinion on pharmacotherapy. 2015 Jun:16(8):1137-47. doi: 10.1517/14656566.2015.1038238. Epub 2015 Apr 20
[PubMed PMID: 25892092]
Level 3 (low-level) evidence
[54]
Ballardie FW, Roberts ISD. Controlled prospective trial of prednisolone and cytotoxics in progressive IgA nephropathy. Journal of the American Society of Nephrology : JASN. 2002 Jan:13(1):142-148. doi: 10.1681/ASN.V131142. Epub
[PubMed PMID: 11752031]
[55]
Pozzi C, Andrulli S, Pani A, Scaini P, Roccatello D, Fogazzi G, Pecchini P, Rustichelli R, Finocchiaro P, Del Vecchio L, Locatelli F. IgA nephropathy with severe chronic renal failure: a randomized controlled trial of corticosteroids and azathioprine. Journal of nephrology. 2013 Jan-Feb:26(1):86-93. doi: 10.5301/jn.5000110. Epub
[PubMed PMID: 22460183]
Level 1 (high-level) evidence
[56]
Barbour SJ, Reich HN. Risk stratification of patients with IgA nephropathy. American journal of kidney diseases : the official journal of the National Kidney Foundation. 2012 Jun:59(6):865-73. doi: 10.1053/j.ajkd.2012.02.326. Epub 2012 Apr 11
[PubMed PMID: 22497792]
[57]
D'Amico G. Natural history of idiopathic IgA nephropathy: role of clinical and histological prognostic factors. American journal of kidney diseases : the official journal of the National Kidney Foundation. 2000 Aug:36(2):227-37
[PubMed PMID: 10922300]