Continuing Education Activity
Prader Willi syndrome (PWS) is a rare and complex genetic disease. The condition has numerous implications on metabolic, endocrine, and neurologic systems. It also presents with behavior and intellectual difficulties as well. PWS is mainly characterized by severe hypotonia with feeding difficulties in the first years of life followed by global developmental delays, hyperphagia, and gradual development of morbid obesity at about three years of age. It is also recognizable by typical facial features, strabismus, and other musculoskeletal conditions. This activity reviews the evaluation and management of Prader Willi Syndrome and highlights the role of the interprofessional clinical team in improving care for patients with the condition.
Objectives:
- Identify the etiology and epidemiology of Prader Willi Syndrome, medical conditions and emergencies.
- Review the appropriate history, physical, and evaluation of Prader Willi Syndrome.
- Describe the treatment and management options available for Prader Willi Syndrome.
- Identify some interprofessional team strategies for improving care coordination and communication to advance Prader Willi Syndrome and improve outcomes.
Introduction
Prader Willi syndrome (PWS) is a rare and complex genetic disease, with numerous implications on metabolic, endocrine, neurologic systems, with behavior and intellectual difficulties. PWS is mainly characterized by severe hypotonia with feeding difficulties in the first years of life. Global developmental delays, hyperphagia with a gradual development of morbid obesity at about three years of age. It is also recognizable by facial features, strabismus, and other musculoskeletal conditions.
Many patients with PWS manifest short stature due to growth hormone deficiency. These individuals also present with hypothalamic dysfunction, leading to several endocrinopathies such as hypogonadism, hypothyroidism, central adrenal insufficiency, with reduced bone mineral density. Therefore these patients need to be closely followed by an endocrinologist throughout their lifespan.[1]
Etiology
Prader Willi Syndrome results from the loss of gene expression within the paternally-inherited genes on the 15q11.2-q13 chromosome results from errors in the genomic imprinting due to paternal deletion in about 70%, maternal uniparental disomy 25% of the cases and defects in the imprinting center such as microdeletions or epimutations on chromosome 15 accounts for the remaining individuals.[1][2] Most cases of Prader Willi syndrome are sporadic, but familial cases can present when the paternal genes carry a microdeletion in the imprinting center inherited from his mother.[2]
Epidemiology
Prader Willi Syndrome has a prevalence of 1 in every 1 in 20000 to 1 in 30000 births.[3]. The mainstay of diagnosis is DNA methylation testing to identify any defect in the parental imprinting on chromosome 15, and this test can detect more than 99% of all affected individuals.[4] In developing countries, diagnosis can be made early in life at around 8.6 weeks of age. Nonetheless, in the majority of cases, confirmation is done at 3.9 years of age, even in centers of reference.[5]
Globally, the number of individuals with Prader Willi Syndrome is approximately 400000, and 20000 of these cases are in the United States.[2]
The most common genetic etiology of life-threatening obesity is Prader Willi syndrome.[2]
Females and males can be equally affected, and there is no difference between races and ethnicities.[6]
Pathophysiology
At birth, growth parameters, such as weight, length, and body mass index, are 15 to 20% smaller in patients with Prader Willi Syndrome than those of their unaffected siblings, indicating that the growth is irregular during the prenatal period. Hypotonia prenatally causes decreased fetal movement, an abnormal position at the time of the delivery leading to increased cesarean and assisted birth.[7]
History and Physical
Clinical Manifestations
Hypotonia:
Infantile hypotonia is very characteristic and found in nearly all individuals with Prader Willi syndrome. They have poor suck reflex producing feeding difficulties, decreased muscle mass, and strength, failure to thrive early in life, requiring to have special equipment for feeding or the need for gavage to improve the nutrition for an extended period.[2][7]
Dysmorphic features:
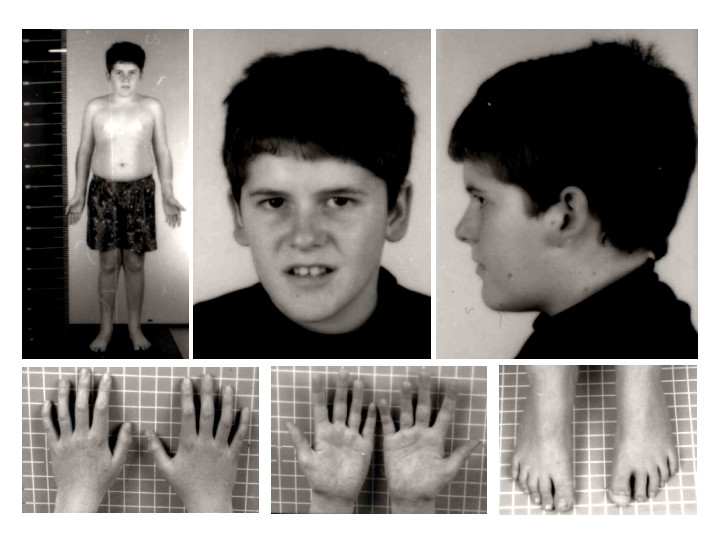
Some facial characteristics include almond-shaped palpebral fissures, narrow frontal diameter, strabismus, narrow nasal bridge, thin upper vermilion with downturned corners of the mouth, and enamel hypoplasia. They can also present with small hands and feet, this could not be apparent at birth, but can evolve slowly over time.[2][7]
Developmental delays and behavioral issues:
Motor developmental delay is present in the majority of individuals with Prader Willi syndrome. Milestone acquisition is, on average, at about double the usual age.[7] Language impairment is also typically seen, intellectual and learning disabilities can fluctuate but are usually more evident at the time the individual reaches school age.[5] Almost all cases of PWS are associated with some degree of behavioral problems, such as anxiety, obsessive-compulsive disorder, temper outbursts, and self-inflicted injuries.[6]
Growth hormone deficiency:
Short stature can be present since early childhood and appears in almost all cases in the second decade of life. Growth hormone deficiency is the most common endocrinopathy reported. Although the pathogenesis is not completely clear, IGF-1 is deficient or subnormal in almost every patient with Prader Willi syndrome. In addition to impairment in the growth hormone secretion test, 24-hour growth hormone spontaneous secretion is also abnormal.[1][7] They present with short stature, growth deceleration, and are not able to exhibit the typical growth spurt during puberty.[8]
Hyperphagia and obesity
There are four nutritional phases described, ranging from failure to thrive to obesity, which is the hallmark of Prader Willi Syndrome. Phase 0: In utero, showing decreased fetal movements and growth restriction. Phase 1: infants are hypotonic and not obese. Divided in a subphase 1a consisting of difficulty feeding with or without failure to thrive from birth to 15 months of age, followed by another subphase 1b characterized by a steady growth at an average rate of weight gain of the infant. Phase 2: the weight gain begins. Split into 2a, where weight increases with no significant changes in calorie intake or appetite; in subphase 2b weight increments now associated with increased appetite and interest for food at a median age of 4.5 years old. Phase 3: represented by hyperphagia related to food-seeking behavior and lack of satiety, the median age of 8 years. Phase 4: some patients reach this phase, where insatiable appetite is no longer present.[2][7]
Hyperphagia that occurs in phase 3, in correlation with the food-seeking behavior, stealing of food or money for food, associated with the decrease of total caloric requirement, results in obesity. In Prader Willi syndrome, obesity is mainly central, in the abdomen, thighs, and buttocks, leading to significant complications with increased morbidity and mortality.[7]
Hypogonadism:
Hypogonadism is present in all patients with Prader Willi syndrome. Cryptorchidism is a feature in almost all male cases, and many of them are required to have orchiopexy. Penile size is reportedly average at birth, but length starts to fall 2 SD under the standard curve later in time. As a result of smaller penile size and increase fatty pad over the pubic area; urination can be troublesome while standing, a short course of testosterone can be helpful in potty-training. At the time of puberty, which appears to occur at an average age, testosterone increases but remains low; generally, LH remains at low-normal levels, while FSH remains normal to high levels. At Tanner stage 3, the arrest of puberty is noticeable, finding testicular failure and a small testicular size that persists into adulthood. Females are born with hypoplasia of external genitalia, puberty usually starts at an adequate age, but breast development delays significantly. Very few patients with PWS can reach menarche and occurs on average at 20 years of age. Estrogen and LH are low to normal, while FSH is normal to hight in the pubertal period, which can be indicative of central and primary defects. Males and females with PWS are considered infertile. There is a high percentage of premature adrenarche in both genders, and estimates are that in about 14 to 30 % of cases, it correlates with advanced bone age, which could be the consequence of obesity.[1]
Other manifestations: Sleep disorders, hypopigmentation, thick viscous saliva, high pain threshold, decreased vomiting, temperature instability, scoliosis/kyphosis, osteoporosis.
Evaluation
Investigations for Prader Willi syndrome should begin with molecular testing with DNA methylation. Chromosome analysis with fluorescence in situ hybridization (FISH) can identify the 15q11-q13. Chromosomal microarray can help identify maternal disomy.[2]
- The thyroid function test is essential to assess hypothyroidism, especially before initiating any growth hormone treatment.[2]
- Liver function tests and serum insulin-like growth factor-1 (IGF-1) to evaluate growth hormone deficiency.[2]
- Fasting glucose levels and oral glucose tolerance test to investigate diabetes.
- Polysomnography (sleep study) to diagnose any sleep disorders.
- Dual X-ray absorptiometry (DXA scan) to assess bone mineralization and composition.[2]
Differential Diagnosis
Neonatal hypotonia is present in several neuropathies and myopathies, such as spinal myotonic dystrophy, which is differentiated by its poor respiratory effort early in life.[7] There are some other syndromes called Prader Willi like Syndrome (PWLS) that share some physical features with Prader Willi syndrome but differs on a genetic basis. Some clinical manifestations of PWLS are hypotonia, short stature, obesity, developmental delays.[9]
Some patients that present with the craniopharyngioma at an early age and the consequences of its treatment at the level of the hypothalamus can manifest with many clinical features that overlap Prader Willi Syndrome.[7]
Prognosis
The prognosis of patients with Prader Willi syndrome will depend on the early clinical course and the degree of complications. Patients managed with early initiation and ongoing treatment could reach an average lifespan, considering that each individual would benefit from life support to achieve as much self-sufficiency as possible.
Complications such as obesity, diabetes, heart failure, is left unresolved can shorten the life expectancy. Death usually occurs in the fourth decade if comorbidities are not well controlled, but if the weight is well regulated, they can reach the seventh decade.[2]
Deterrence and Patient Education
Pediatricians need to try to diagnose Prader Willi Syndrome as early as possible. Early diagnosis can address many problems during infancy, and this could increment the possibility of initiating treatment at a young age leading to better outcomes.
Enhancing Healthcare Team Outcomes
Prader Willi syndrome is a complex genetic disorder that is caused by a defect in chromosome 15. Pediatricians need to suspect this diagnosis when a neonate demonstrates hypotonia because it can lead to several complications, including difficulty feeding and failure to thrive.
Following the diagnosis, it is crucial to incorporate an interprofessional team such as geneticist, endocrinologist, developmental specialist, a nutritionist that can provide an integrated approach, and they will monitor disease progression, complications. They will evaluate the need for early initiation of therapy to achieve better outcomes. A board-certified clinical pharmacist should be on board for the dosing of hormonal replacement and augmentation. Nursing staff can monitor and record the patient's response to therapy at each visit, charting results so the clinical team can make modifications to the regimen as needed. A mental health nurse may also be a crucial component of an overall interprofessional care plan. This interprofessional team approach is necessary to optimize outcomes, patient quality of life, and longevity in patients with Prader Willi syndrome. [Level 5]