Introduction
Amegakaryocytic thrombocytopenia in both congenital and acquired forms represents a severe subtype of thrombocytopenia characterized by diminished or absent megakaryocytes without additional bone marrow abnormalities.[1] Congenital amegakaryocytic thrombocytopenia manifests with severe thrombocytopenia and various bleeding manifestations at birth, often progressing to bone marrow failure. In contrast, acquired amegakaryocytic thrombocytopenia typically emerges later in life, possibly due to immune-mediated mechanisms.[2][3][4][5][6][7][8] Acquired amegakaryocytic thrombocytopenia is typically a diagnosis of exclusion, becoming apparent as bleeding complications following treatment failure of immune thrombocytopenia.
Diagnosis involves bone marrow biopsy and genetic testing. Clinicians diagnose congenital amegakaryocytic thrombocytopenia by demonstrating homozygous or compound heterozygous mutations in the c-myeloproliferative leukemia virus, thrombopoietin receptor (c-MPL) proto-oncogene. In contrast, clinicians base the diagnosis of acquired amegakaryocytic thrombocytopenia on clinical suspicion and reduced or absent megakaryocytes in the bone marrow. Allogeneic hematopoietic stem cell transplant is the only curative option for patients with congenital amegakaryocytic thrombocytopenia and a c-MPL mutation, whereas treating the underlying condition combined with immunosuppressive medications targets acquired amegakaryocytic thrombocytopenia.
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
Congenital Amegakaryocytic Thrombocytopenia
Congenital amegakaryocytic thrombocytopenia is inherited in an autosomal recessive manner, typically arising from deficient thrombopoietin signaling due to a biallelic mutation of c-MPL on chromosome 1p34.[9] This gene encodes the thrombopoietin (THPO) or myeloproliferative leukemia virus (MPL) receptor. Researchers have identified nearly 41 mutations within c-MPL associated with congenital amegakaryocytic thrombocytopenia to date.[5][9][5][10] There are two primary forms of congenital amegakaryocytic thrombocytopenia, as described by experts.[5]
Type 1: Congenital amegakaryocytic thrombocytopenia-1 (OMIM 604498) arises from a homozygous or compound heterozygous mutation in c-MPL, which causes a complete loss of the THPO receptor, abolishing receptor signaling.[11] Consequently, affected individuals experience severe thrombocytopenia at birth, with an elevated susceptibility to intracranial bleeding and an early onset of bone marrow failure, typically occurring by 33 months.[4][10][11][12]
Type 2: Congenital amegakaryocytic thrombocytopenia-2 arises due to either a splicing anomaly or an amino acid substitution, impacting glycosylation of the MPL receptor and consequently hampering its responsiveness to THPO. Such mutations may further disrupt hydrogen bonds within the MPL receptor, leading to receptor instability, albeit leaving some residual receptor functions intact. Children with congenital amegakaryocytic thrombocytopenia-2 have mild thrombocytopenia that transiently normalizes during the first year of life. The rate of bone marrow failure in congenital amegakaryocytic thrombocytopenia-2 is slower compared to that in congenital amegakaryocytic thrombocytopenia-1, typically occurring in patients aged 3 to 6, with a mean age of 5.[11][12]
Although a biallelic variation in c-MPL occurs in most patients with congenital amegakaryocytic thrombocytopenia-2, additional genetic variations are possible. A homozygous variation of THPO (OMIM 600044.0011) prevents liver cells from producing THPO.[13][14] According to Thompson and Nguyen, abnormalities in the homeobox A11 (HOXA11) and MDS1 and EVI1 complex locus (MECOM) genes lead to congenital amegakaryocytosis, thrombocytopenia, and eventual bone marrow failure in most patients.[3][15] Clinicians differentiate patients with the HOXA11 and MECOM genetic variations from other forms of congenital amegakaryocytic thrombocytopenia due to their association with radioulnar synostosis. MECOM-associated syndrome is an autosomal dominant form of congenital amegakaryocytic thrombocytopenia, also presenting with bone marrow failure, radioulnar synostosis, B-cell deficiency, and cardiorenal dysfunction.[16] Mutations in this gene affect factor EVI1, which controls MPL transcription. In addition, a null mutation in RBM8A leads to thrombocytopenia-absent radius syndrome, which is differentiated from other forms of congenital amegakaryocytic thrombocytopenia by the presence of bilateral radial aplasia. Additional proposed alternate etiologies include an X-linked variety of congenital amegakaryocytic thrombocytopenia and an anti-human leukocyte antigen (HLA) A2 antibody.[10][13][17]
Acquired Amegakaryocytic Thrombocytopenia
The exact mechanism of acquired amegakaryocytic thrombocytopenia is unknown. Researchers have proposed 3 potential mechanisms.[18][19]
Suppression of megakaryocyte maturation by an exogenous agent: Ebstein-Barr virus, parvovirus B19, hepatitis C virus, interferon therapy, cytomegalovirus, benzene exposure, alcohol use disorder, vitamin B12 deficiency, and radioiodine therapy are all associated with acquired amegakaryocytic thrombocytopenia.[20][21]
Suppression of megakaryocyte maturation by endogenous stimuli due to antibody-mediated or T-cell autoimmunity: Researchers proposed dysregulated humoral immunity as one of the mechanisms for acquired amegakaryocytic thrombocytopenia due to the presence of anti-THPO immunoglobulin G antibodies and autoantibodies against the THPO receptor, which block the function of THPO. AAMT has been observed in correlation with thymoma, manifesting with a more aggressive disease progression. Additional associations are adult-onset Still disease, eosinophilic fasciitis, systemic lupus erythematosus, autoimmune hemolytic anemia, systemic sclerosis, Graves' disease, and hyperestrogenic states.[18][22][23][24] The response of acquired amegakaryocytic thrombocytopenia to immunosuppressant medication lends further support to immune-mediated pathogenesis.
An early manifestation of a stem cell abnormality: Acquired amegakaryocytic thrombocytopenia is a precursor to various conditions, including acute myeloid leukemia, myelodysplastic syndrome, aplastic anemia, and non-Hodgkin lymphoma.[7][25] Other reports associate acquired amegakaryocytic thrombocytopenia with large granular lymphocyte leukemia, and the Philadelphia chromosome and 5q deletion.[25][26] These observations indicate that the defect in acquired amegakaryocytic thrombocytopenia originates in an early progenitor cell within the megakaryocytic lineage.
Epidemiology
With fewer than 100 reported cases, congenital amegakaryocytic thrombocytopenia is considered rare.[10] Consanguinity is suggested as a potential factor, and there appears to be a slight female predominance.[5] The incidence is likely underestimated, possibly due to misdiagnosis as neonatal alloimmune thrombocytopenia. Differentiating congenital amegakaryocytic thrombocytopenia from primary aplastic anemia can be challenging, particularly once the patient has advanced to pancytopenia.[27]
Likewise, the actual incidence of acquired amegakaryocytic thrombocytopenia is probably higher compared to that reported, as clinicians may diagnose many cases as immune thrombocytopenia.[25] Females affected by acquired amegakaryocytic thrombocytopenia typically receive their diagnosis between the ages of 40 and 60. In contrast, most affected males are at either end of the age spectrum, with peaking when they are aged 60.[28]
Pathophysiology
Congenital Amegakaryocytic Thrombocytopenia
Megakaryopoiesis initiates from the hematopoietic stem cell within the bone marrow. The hematopoietic stem cell undergoes maturation into a multipotent progenitor cell, transitioning into a committed megakaryocyte progenitor cell, then an immature megakaryocyte, and ultimately matures into a fully developed megakaryocyte, responsible for generating platelets in the blood.[29] Notably, THPO acts as a crucial cytokine during the process of megakaryopoiesis, facilitating the proliferation, enlargement, and polyploidy of megakaryocytes while stimulating the expression of platelet-specific markers.[4] In addition, THPO boosts the expression of vascular endothelial growth factor, homeobox B4 (HOXB4), and homeobox A9 (HOXA9), thereby supporting the growth and survival of hematopoietic stem cells.[10] Although the liver primarily synthesizes THPO, the body can also produce some in the proximal tubular cells of the kidneys and bone marrow stromal cells.[30]
Located in the bone marrow, liver, spleen, and on CD34+ cells, THPO binding initiates a cascade of signaling events within the target cell through the Janus kinase/signal transducers and activators of transcription kinase proteins, mitogen-activated protein kinase, and phosphatidylinositol-3-kinase pathways.[4][30] MPL signaling is vital for differentiating multipotent progenitor cells into megakaryocyte and erythrocyte progenitor cells. Problems in signaling can lead to impaired megakaryocyte and erythrocyte production, as noted in the later stages of congenital amegakaryocytic thrombocytopenia.[31] Notably, decreased expression of P-selectin on neonatal platelets can reduce platelet activation and predispose affected infants to the high bleeding tendency pre- or perinatally in patients with congenital amegakaryocytic thrombocytopenia.[32]
Acquired Amegakaryocytic Thrombocytopenia
Antibody or T-cell–mediated autoimmunity may cause acquired amegakaryocytic thrombocytopenia.[21] The proposed pathophysiology includes antibodies targeted against THPO, antigens on megakaryocyte progenitor cells, granulocyte-monocyte colony-stimulating factor, or the megakaryocyte colony-forming unit. Failure of terminal megakaryocyte differentiation, suppression of megakaryocyte colony-forming unit by T cells and adherent monocytes, monoclonal T-cell population destroying megakaryocyte lineage, or a defect in cytokine-mediated regulation of megakaryopoiesis are additional proposed mechanisms.[6][22][25][33]
Systemic lupus erythematosus and systemic sclerosis have shown associations with antibodies against the MPL receptor.[21][34] In cases of hepatitis C infection, anti-MPL antibodies are produced. Initially, these antibodies bind to MPL receptors on platelets, leading to their destruction in the spleen, mimicking the clinical presentation of immune thrombocytopenia. Later, these antibodies bind to the MPL receptor on megakaryocytes, blocking the function of THPO and resulting in acquired amegakaryocytic thrombocytopenia.[35] Interestingly, interferon therapy used to treat patients with hepatitis C can also generate anti-MPL antibodies.[35]
Histopathology
The bone marrow evaluation in patients with amegakaryocytic thrombocytopenia typically demonstrates normal overall cellularity with a reduction or absence of megakaryocytes. The megakaryocytes may look immature or small, with no evidence of dysplasia.[4][10][11] Immunohistochemical staining for CD-61 megakaryocyte antigen shows diminished megakaryocytes on core biopsy (see Image. Bone Marrow Core Biopsy).[7]
Patients with congenital amegakaryocytic thrombocytopenia may have minimal findings on bone marrow evaluation early in the disease, making the diagnosis challenging. Serial bone marrow biopsies may be required to confirm the diagnosis.[11] On peripheral smear, platelets typically exhibit normal size and morphology.[3][11] Later, once pancytopenia develops, affected patients have hypocellular marrow with decreased progenitor cells in all lineages, making it challenging to distinguish congenital amegakaryocytic thrombocytopenia from other causes of aplastic anemia.[10]
History and Physical
Thrombocytopenia is a platelet count <150,000 cells/µL. Patients with congenital amegakaryocytic thrombocytopenia typically exhibit severe thrombocytopenia, often with a platelet count of less than 21,000 cells/μL, presenting either at birth or within the initial month of life. Occasionally, affected infants develop findings during fetal development. Common manifestations include purpura, intracranial bleeding, recurrent rectal bleeding, or pulmonary hemorrhage within hours of birth.[4][10] A family history of thrombocytopenia may be present.[4] No characteristic congenital phenotypic abnormalities are associated with congenital amegakaryocytic thrombocytopenia. Some studies note neurological defects such as strabismus, cerebellar agenesis, hypoplasia of the corpus callosum and brainstem, facial malformations, and cortical dysplasia. The underlying mechanism of these neurological findings is unclear. One possibility is that the absence or deficiency of MPL in the brain, as observed in congenital amegakaryocytic thrombocytopenia, can lead to developmental delay. The other hypothesis considers the long-term sequelae due to intracranial bleeding.[3][5][10][11][36]
Acquired amegakaryocytic thrombocytopenia is a diagnosis of exclusion, and the diagnosis often becomes apparent when patients develop bleeding complications after failing standard treatment for immune thrombocytopenia, including steroids or intravenous immunoglobulin therapy.[7] Patients with acquired amegakaryocytic thrombocytopenia can present with petechiae, purpura, ecchymosis, easy bruising, epistaxis, or fatigue. Splenomegaly is absent.[8] A case report describes a patient with acquired amegakaryocytic thrombocytopenia presenting with massive hemoperitoneum due to a hemorrhagic corpus luteum.[37]
Evaluation
General Evaluation
When evaluating a child with thrombocytopenia, clinicians should consider the age of onset and systemic symptoms such as fever, bone pain, changes in appetite, weight loss, and decreased energy, which may indicate an underlying systemic disease such as malignancy or autoimmune disorders. In such cases, prompt evaluation is essential due to these conditions' severe potential morbidity and mortality. Reviewing previous blood counts, if available, is crucial. A history of thrombocytopenia suggests a possible congenital or chronic disorder, whereas normal platelet counts in the past may indicate an acquired condition.
Clinicians should consider prodromal illnesses, as triggers such as viral infections or vaccinations can trigger immune thrombocytopenia. Symptoms such as abdominal pain and bloody diarrhea may suggest Shiga-toxin-associated hemolytic uremic syndrome, particularly if occurring following exposure to enteric pathogens. A thorough review of medication history is necessary, as certain medications, including chemotherapeutic agents, heparin, antiseizure medications, and antibiotics, may lower platelet counts through various mechanisms.
During the physical examination of children presenting with thrombocytopenia, clinicians should meticulously evaluate and document any bleeding manifestations, especially those in the skin. This examination includes careful observation of petechiae, nonpalpable purpura, and ecchymoses, with particular emphasis on noting bleeding sites, especially in dependent areas of the body. Serial examinations are crucial in tracking changes in bleeding patterns or the number of petechiae over affected areas. In hospitalized patients, thorough examination for signs of bleeding is essential, with attention paid to catheter sites, drains, incisions, areas of previous trauma, and exit sites of venous access devices. In addition, a comprehensive assessment of the gingival and oral cavity is indispensable to identify any evidence of bleeding.
Various aspects of the physical examination can offer valuable insights into the underlying cause of thrombocytopenia. Short stature may signal inherited bone marrow failure syndromes, whereas congenital anomalies such as radial and thumb abnormalities or cleft palate may suggest specific inherited thrombocytopenia syndromes. Neurological symptoms such as confusion, headache, stroke, or seizures may indicate conditions such as thrombotic thrombocytopenic purpura. Lymphadenopathy might point towards underlying lymphoid malignancies or infectious causes of thrombocytopenia. Moreover, specific features such as hearing loss, cataracts, and leukoplakia may be associated with certain inherited platelet disorders. Abdominal examination may unveil splenomegaly, which can arise from various etiologies such as viral infections, leukemia, or chronic liver disease. Joint swelling could indicate autoimmune diseases such as systemic lupus erythematosus, where thrombocytopenia may manifest initially. Furthermore, skeletal abnormalities such as absent radii or thumb abnormalities may hint at specific syndromes linked with thrombocytopenia. Finally, cutaneous findings such as eczema, pigmentary changes, café-au-lait spots, or vascular tumors may also offer diagnostic clues.
Laboratory Evaluation
In addition to a platelet count, a complete blood count and peripheral blood smear are essential to assess all blood cell indices, platelet size, morphology characterization, and evidence of platelet clumping or abnormalities in red and white cells. Bone marrow biopsy is the mainstay of diagnosing congenital amegakaryocytic thrombocytopenia or acquired amegakaryocytic thrombocytopenia.
Congenital amegakaryocytic thrombocytopenia
Bone marrow biopsy is warranted for all children with severe thrombocytopenia or platelet counts <50,000 cells/μL at birth and platelets of normal size and morphology. Clinicians perform c-MPL gene testing when a reduced number of megakaryocytes are present on the biopsy.[27]
The c-MPL gene analysis is performed with bidirectional sequencing of all 12 exons, including coding regions, splice sites, and intron-exon boundaries. Clinicians obtain samples from whole blood, bone marrow, skin fibroblasts, buccal brushings, amniotic fluid, or CD34+ cells.[4][38] Chromosome analysis and fluorescence in situ hybridization studies are alternative genetic tests.
The presence of either homozygous or compound heterozygous mutations c-MPL confirms the diagnosis of congenital amegakaryocytic thrombocytopenia.[5] Plasma THPO levels are elevated. THPO levels rise 10-fold as THPO internalization and destruction by MPL receptors do not occur. Patients should also be screened for THPO mutations because congenital amegakaryocytic thrombocytopenia caused by these mutations can be treated with an MPL agonist such as romiplostim.[13] In patients with THPO mutations, the levels of THPO are low due to a defect in the secretion of THPO.[11][13]
Acquired amegakaryocytic thrombocytopenia
A strong clinical suspicion for acquired amegakaryocytic thrombocytopenia is necessary. As with congenital amegakaryocytic thrombocytopenia, patients with suspected acquired amegakaryocytic thrombocytopenia require a complete blood count and peripheral smear. Hepatic enzymes, coagulation testing, and albumin levels are essential to evaluate liver function. HIV and hepatitis C virus testing should also be incorporated. The patient's presentation and physical examination findings dictate further testing. Antinuclear or antiphospholipid antibodies may be necessary in patients with systemic lupus erythematosus or antiphospholipid syndrome indicators. Clinicians should consider cultures, prothrombin time, activated partial thromboplastin time, and fibrinogen to evaluate for sepsis or disseminated intravascular coagulation in patients with a fever or evidence of systemic infection. Patients with microangiopathic changes on the peripheral smear should undergo coagulation testing, lactate dehydrogenase, and creatinine to evaluate for disseminated intravascular coagulation, thrombotic thrombocytopenic purpura, or hemolytic uremic syndrome. The bone marrow biopsy reveals an isolated reduction or absence of megakaryocytes in patients with acquired amegakaryocytic thrombocytopenia.
Treatment / Management
Congenital Amegakaryocytic Thrombocytopenia
Allogeneic hematopoietic stem cell transplant is the sole curative treatment option for individuals with congenital amegakaryocytic thrombocytopenia and a c-MPL mutation.[10] Clinicians should consider hematopoietic stem cell transplant early in the disease course, before the onset of pancytopenia, due to the risks associated with multiple transfusions, alloimmunization, and infection.[10][27] Some research suggests performing HLA typing for patients and their siblings at diagnosis. A case report illustrates a successful transplant from a patient's father, who served as a haploidentical donor with 5 out of 10 HLA-matched alleles despite carrying the c-MPL variation.[12] The typical age for hematopoietic stem cell transplant is approximately 38 months, ranging from 7 to 89 months.[10] (B3)
Hematopoietic stem cell transplant's conditioning regimens include busulfan, cyclophosphamide, and total body irradiation.[5] These regimens are fully myeloablative and carry survival rates of approximately 80% but often cause significant pulmonary, mucosal, and hepatic toxicity.[39] Infertility is a potential long-term result, and discussions regarding future fertility and fertility preservation are necessary. For HLA-mismatched hematopoietic stem cell transplant, the 1-year incidence of graft failure is 19%, whereas for HLA-matched hematopoietic stem cell transplant, the incidence is 7%.[40](B3)
Recent developments reveal encouraging outcomes with the utilization of non-myeloablative protocols.[25][39] These treatment regimens, similar to those utilized in managing acquired aplastic anemia with antithymocyte globulin therapy, demonstrate advantages such as fertility preservation, reduced overall toxicity, and successful engraftment of myeloid chimerism.(B3)
Supportive treatment includes irradiated, leukocyte-reduced platelet transfusions; antifibrinolytics such as tranexamic acid; and avoidance of nonsteroidal anti-inflammatory medications and aspirin. Once pancytopenia develops, packed red blood cells and antibiotics may be required.
The rarity of this disease thwarts the creation of clinical trials. Hematopoietic stem cell transplant does not benefit patients with a THPO variation since the liver produces THPO.[14] Romiplostim, a THPO peptide mimetic, and eltrombopag, a small molecule agonist of the MPL receptor that induces conformational changes in the receptor, have proven beneficial for patients with congenital amegakaryocytic thrombocytopenia due to THPO variations.[13][30]
Acquired Amegakaryocytic Thrombocytopenia
Currently, no established treatment guidelines exist for acquired amegakaryocytic thrombocytopenia. The goal of therapy is to treat the underlying etiology.[41] Hoffman suggests using plasmapheresis, cyclophosphamide, cyclosporine, and prednisone for antibody-mediated acquired amegakaryocytic thrombocytopenia and cyclosporine, cytokine therapy, and antithymocyte globulin for T-cell–mediated acquired amegakaryocytic thrombocytopenia.[42] Some experts advised starting with the least toxic and most cost-effective medications with periodic treatment response monitoring.[37] The following drugs and therapies have been used for the treatment of AMMT with varying degrees of success.(B3)
- Corticosteroids suppress immune B- and T-cell–mediated autoimmunity.
- High-dose intravenous immunoglobulin therapy binds antibodies against THPO and megakaryocytes.
- Rituximab suppresses the production of autoantibodies by B cells.
- Cyclosporine.
- Antithymocyte globulin suppresses T-cell–mediated autoimmunity and stimulates hematopoiesis directly.
- Bone marrow transplant.
- Cyclophosphamide or azathioprine.
- Lithium carbonate, vincristine, mycophenolate mofetil, danazol, and eltrombopag.
- Recombinant interleukin-11 proteins.[43] (B3)
Corticosteroids and intravenous immunoglobulin are frequently inadequate or provide only transient initial improvement. Cyclosporine monotherapy is often effective. Clinicians consider bone marrow transplantation in patients who are refractory to treatment with cyclosporine or antithymocyte globulin or those who have progressed to aplastic anemia or myelodysplastic syndrome. A case report describes the successful treatment of adult-onset Still disease with tocilizumab, a humanized anti-interleukin 6 receptor antibody and cyclosporine.[21] Some refractory cases of acquired amegakaryocytic thrombocytopenia have been treated with a combination of antithymocyte globulin and cyclosporine.[25][33][44] Other reports reveal a positive response to avatrombopag, a THPO receptor agonist, despite being refractory to eltrombopag.[45](B3)
Differential Diagnosis
Congenital Amegakaryocytic Thrombocytopenia
The severity of thrombocytopenia, age at onset, and maternal history are all factors to consider when developing differential diagnoses for neonates and infants with thrombocytopenia. The following list contains the differential diagnoses for congenital amegakaryocytic thrombocytopenia:
- Birth asphyxia or placental insufficiency;
- Congenital toxoplasmosis, rubella, cytomegalovirus, and herpes simplex virus infections;
- Sepsis;
- Congenital syphilis;
- Varicella;
- Parvovirus B19;
- Drug-induced thrombocytopenia due to drug-dependent antibodies;
- Disseminated intravascular coagulation;
- Thrombosis;
- Type 2B von Willebrand disease;
- Hypersplenism due to hemolytic anemia, congenital hepatitis due to viral infection, and portal vein thrombosis;
- Wiskott-Aldrich syndrome;
- Fanconi anemia;
- Preeclampsia;
- Trisomies 21, 18, and 13;
- Turner syndrome;
- Neonatal leukemia or neuroblastoma;
- Neonatal autoimmune thrombocytopenia due to maternal antibodies that react with both maternal and fetal platelets in systemic illnesses such as systemic lupus erythematosus;
- Neonatal alloimmune thrombocytopenia due to fetal platelets containing an antigen inherited from the father that the mother lacks;
- Kasabach-Merritt syndrome; and
- Dyskeratosis congenita.[4][5][10][11][32]
Acquired Amegakaryocytic Thrombocytopenia
The etiology of thrombocytopenia in adults is extensive.[43] Most commonly, acquired amegakaryocytic thrombocytopenia is diagnosed as immune thrombocytopenia. The typical presentation is a patient with immune thrombocytopenia who fails to improve with corticosteroids or intravenous immunoglobulin. Additional potential diagnoses are as follows:
- Liver disease;
- Antiplatelet autoantibodies induced by vaccines, foods, or other substances;
- Post-transfusion purpura;
- Thrombosis;
- Disseminated intravascular coagulation;
- Postoperative thrombocytopenia;
- Massive fluid resuscitation or massive transfusion;
- Gestational thrombocytopenia;
- Infections, such as Epstein–Barr virus, varicella, leptospirosis, anaplasmosis, dengue, babesiosis, and tick-borne diseases;
- Medications, including daptomycin, valproic acid, linezolid, and penicillin;
- Beverages, including alcohol, tonic water containing quinine, and herbal supplements;
- Vitamin B12, copper, and folic acid deficiency;
- Paroxysmal nocturnal hemoglobinuria;
- Hematologic cancers or solid organ cancers with bone metastasis;
- Chemotherapy and radiation therapy for hematologic cancers or metastatic cancer;
- Rheumatologic conditions, such as systemic lupus erythematosus with or without antiphospholipid syndrome;
- Peripheral destruction of platelets by shearing stress as observed in the use of intra-aortic balloon pumps and aortic aneurysms;
- Splenic sequestration of platelets;
- Aplastic anemia and other bone marrow failure syndromes; and
- Consumptive coagulopathies such as disseminated intravascular coagulation, heparin-induced thrombocytopenia with thrombosis, or hemolytic uremic syndrome.[46]
Pertinent Studies and Ongoing Trials
Experimental treatments encompass gene therapy utilizing lentiviral vectors, clustered regularly interspaced palindromic repeats (CRISPR) and CRISPR-associated protein 9 (Cas9) gene editing, and investigational medications such as LGD-4665, anti-c-MPL minibodies, and diabodies. Researchers attempt to utilize lentiviral vector gene therapy to rectify c-Mpl variations; however, concerns persist regarding the potential leukemogenicity associated with this method.[10] Others are attempting to use CRISPR-Cas9 gene editing to correct the mutation.[47] Furthermore, LGD-4665, minibodies, and diabodies represent experimental pharmaceuticals that stimulate some variants of MPL receptors to enhance megakaryocyte proliferation.[30][48]
Prognosis
Once pancytopenia develops in patients with congenital amegakaryocytic thrombocytopenia, the prognosis becomes poor. A study reveals a mortality rate of 30% due to bleeding-related complications, with an additional 20% of patients dying due to complications related to hematopoietic stem cell transplant.[4] Approximately 50% of affected patients develop aplastic anemia within the first year of life. Reports also indicate the possibility of leukemia and myelodysplasia later in childhood.[39]
The prognosis and clinical course of acquired amegakaryocytic thrombocytopenia are variable. Some patients may achieve and maintain a prolonged remission, whereas others develop a long course of relapses and remissions. In addition, some may rapidly progress and develop aplastic anemia, myelodysplastic syndrome, or leukemia despite therapy. Patients who develop aplastic anemia have a poor prognosis.[7][8]
Complications
The following list includes potential complications related to acquired amegakaryocytic thrombocytopenia and congenital amegakaryocytic thrombocytopenia:
Deterrence and Patient Education
Amegakaryocytic thrombocytopenia presents a rare but serious threat, underscoring the importance of early diagnosis and intervention to prevent progression to bone marrow failure. Patients and caregivers must grasp the potential risks involved, encompassing hemorrhage, advancement to aplastic anemia, myelodysplastic syndrome, and acute myeloid leukemia. Patients must recognize that achieving a definitive diagnosis is invasive and requires a bone marrow biopsy.
Moreover, treatments for amegakaryocytic thrombocytopenia, particularly congenital amegakaryocytic thrombocytopenia, may entail invasive procedures such as hematopoietic stem cell transplant. Patients and caregivers must be fully informed about the potential hazards and benefits associated with hematopoietic stem cell transplant, stressing the significance of adhering to treatment regimens and understanding the importance of contacting their clinician for evidence of bleeding or infection. In addition, patients and caregivers should be aware of the importance of avoiding activities such as skiing, mountain climbing, and contact sports that heighten the risk of bleeding and intracranial hemorrhage.
Clinicians should discuss the complexities of treatment associated with acquired amegakaryocytic thrombocytopenia, acknowledging the potential for multiple ineffective immunosuppressive and immunomodulatory agents. Patients should know the potential necessity for supportive platelet transfusions and the attendant risks. Ultimately, patient education and awareness are pivotal in managing amegakaryocytic thrombocytopenia, ensuring that patients and caregivers have the knowledge and understanding essential for informed decision-making and risk mitigation.
Enhancing Healthcare Team Outcomes
Amegakaryocytic thrombocytopenia represents a severe form of thrombocytopenia distinguished by reduced or absent megakaryocytes in the bone marrow without additional abnormalities. Managing this condition necessitates a comprehensive, multidisciplinary approach to ensure patient-centered care. Clinically, amegakaryocytic thrombocytopenia includes both congenital and acquired forms. Congenital amegakaryocytic thrombocytopenia typically manifests with profound thrombocytopenia and bleeding issues at birth, often progressing to bone marrow failure. Acquired amegakaryocytic thrombocytopenia typically appears later in life and is potentially linked to immune-mediated mechanisms.
Severe thrombocytopenia and bleeding, at birth or within the initial month of life, represent hallmark clinical features observed in patients with congenital amegakaryocytic thrombocytopenia. Diagnosis typically involves conducting a bone marrow biopsy and genetic testing. Conversely, acquired amegakaryocytic thrombocytopenia often manifests when patients experience bleeding complications after the failure of standard treatments for immune thrombocytopenia. An extensive evaluation, encompassing a review of systemic symptoms, a focused physical examination targeting bleeding manifestations, and laboratory assessments, plays a crucial role in diagnosis and management decisions. Allogeneic hematopoietic stem cell transplant is the only curative approach for patients with congenital amegakaryocytic thrombocytopenia and a c-MPL genetic variation. Conversely, treatment strategies for individuals with acquired amegakaryocytic thrombocytopenia primarily revolve around addressing the underlying cause, often in conjunction with immunosuppressive medications or bone marrow transplantation.
Physicians, advanced practitioners, nurses, pharmacists, and other healthcare professionals contribute unique skills and expertise to the care team. Clinicians must possess the necessary expertise in diagnosis and disease management to guide decision-making, ensuring patients receive a timely diagnosis and appropriate interventions tailored to their needs. Pharmacists play a critical role in medication management, ensuring safe and effective use of pharmacotherapy. Their expertise in drug interactions, dosage adjustments, and adverse effect management optimizes medication regimens, minimizing the risk of complications. Nurses are instrumental in delivering direct patient care, administering medications, and advocating for patients' well-being. Their vigilance in monitoring for complications and adherence to safety protocols contribute to patient safety and positive outcomes.
Other health professionals, such as social workers and genetic counselors, address psychosocial needs and provide genetic education and counseling to patients and families affected by amegakaryocytic thrombocytopenia. Effective interprofessional communication and care coordination among all team members are essential to ensure seamless transitions of care, timely interventions, and holistic support for patients. By leveraging the interdisciplinary team's collective skills, strategy, and expertise, healthcare professionals can deliver comprehensive, patient-centered care that maximizes outcomes, promotes patient safety, and enhances the healthcare team's performance in managing amegakaryocytic thrombocytopenia.
Media
(Click Image to Enlarge)
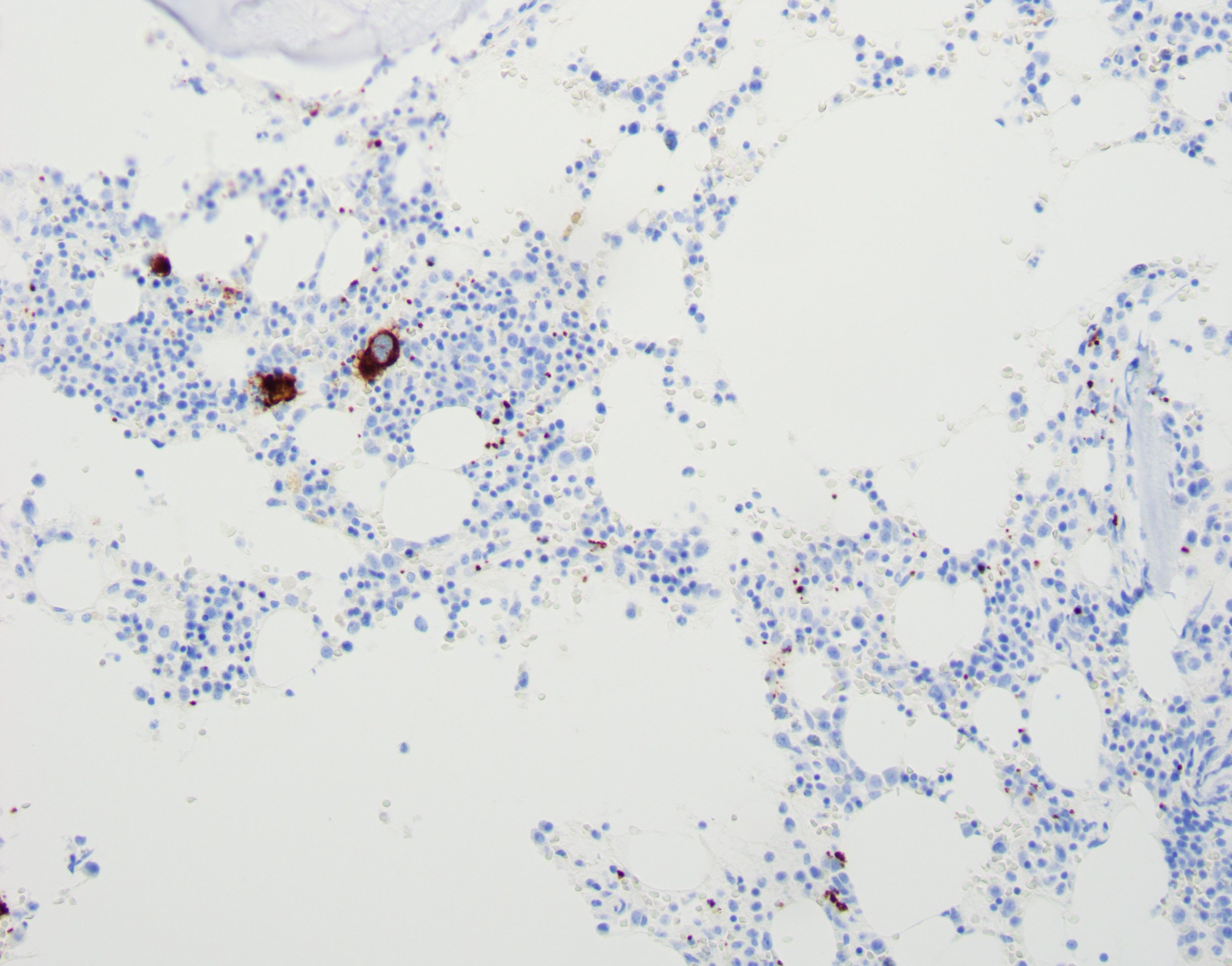
Bone Marrow Core Biopsy. The image depicts CD61 immunohistochemical staining on a bone marrow core biopsy sample from a patient with acquired amegakaryocytic thrombocytopenia, with a decreased number of megakaryocytes highlighted in brown.
Department of Pathology, Rochester General Hospital, New York
References
Balduini CL. The name counts: the case of 'congenital amegakaryocytic thrombocytopenia'. Haematologica. 2023 May 1:108(5):1216-1219. doi: 10.3324/haematol.2022.282024. Epub 2023 May 1 [PubMed PMID: 36226496]
Level 3 (low-level) evidenceMaserati E, Panarello C, Morerio C, Valli R, Pressato B, Patitucci F, Tassano E, Di Cesare-Merlone A, Cugno C, Balduini CL, Lo Curto F, Dufour C, Locatelli F, Pasquali F. Clonal chromosome anomalies and propensity to myeloid malignancies in congenital amegakaryocytic thrombocytopenia (OMIM 604498). Haematologica. 2008 Aug:93(8):1271-3. doi: 10.3324/haematol.12748. Epub 2008 Jun 2 [PubMed PMID: 18519517]
Level 3 (low-level) evidenceKhincha PP, Savage SA. Neonatal manifestations of inherited bone marrow failure syndromes. Seminars in fetal & neonatal medicine. 2016 Feb:21(1):57-65. doi: 10.1016/j.siny.2015.12.003. Epub 2015 Dec 24 [PubMed PMID: 26724991]
Al-Qahtani FS. Congenital amegakaryocytic thrombocytopenia: a brief review of the literature. Clinical medicine insights. Pathology. 2010 Jun 4:3():25-30 [PubMed PMID: 21151552]
Ballmaier M, Germeshausen M. Congenital amegakaryocytic thrombocytopenia: clinical presentation, diagnosis, and treatment. Seminars in thrombosis and hemostasis. 2011 Sep:37(6):673-81. doi: 10.1055/s-0031-1291377. Epub 2011 Nov 18 [PubMed PMID: 22102270]
Simkins A, Maiti A, Short NJ, Jain N, Popat U, Patel KP, Oo TH. Acquired amegakaryocytic thrombocytopenia and red cell aplasia in a patient with thymoma progressing to aplastic anemia successfully treated with allogenic stem cell transplantation. Hematology/oncology and stem cell therapy. 2019 Jun:12(2):115-118. doi: 10.1016/j.hemonc.2017.09.001. Epub 2018 Jan 31 [PubMed PMID: 29409729]
Brown GE, Babiker HM, Cantu CL, Yeager AM, Krishnadasan R. "Almost bleeding to death": the conundrum of acquired amegakaryocytic thrombocytopenia. Case reports in hematology. 2014:2014():806541. doi: 10.1155/2014/806541. Epub 2014 Feb 6 [PubMed PMID: 24649385]
Chaudhary UB, Eberwine SF, Hege KM. Acquired amegakaryocytic thrombocytopenia purpura and eosinophilic fasciitis: a long relapsing and remitting course. American journal of hematology. 2004 Mar:75(3):146-50 [PubMed PMID: 14978695]
Level 3 (low-level) evidenceOk Bozkaya İ, Yaralı N, Işık P, Ünsal Saç R, Tavil B, Tunç B. Severe Clinical Course in a Patient with Congenital Amegakaryocytic Thrombocytopenia Due to a Missense Mutation of the c-MPL Gene. Turkish journal of haematology : official journal of Turkish Society of Haematology. 2015 Jun:32(2):172-4. doi: 10.4274/tjh.2013.0191. Epub [PubMed PMID: 26316487]
Geddis AE. Congenital amegakaryocytic thrombocytopenia. Pediatric blood & cancer. 2011 Aug:57(2):199-203. doi: 10.1002/pbc.22927. Epub 2011 Feb 18 [PubMed PMID: 21337678]
Geddis AE. Congenital amegakaryocytic thrombocytopenia and thrombocytopenia with absent radii. Hematology/oncology clinics of North America. 2009 Apr:23(2):321-31. doi: 10.1016/j.hoc.2009.01.012. Epub [PubMed PMID: 19327586]
Wang S, Yang X, Ai Y, Zhu Y. Haploidentical Hematopoietic Stem Cell Transplantation in a 3-Year-Old Girl with Congenital Amegakaryocytic Thrombocytopenia: A Case Report. Klinische Padiatrie. 2022 Nov:234(6):388-390. doi: 10.1055/a-1933-2583. Epub 2022 Nov 15 [PubMed PMID: 36379227]
Level 3 (low-level) evidenceVarghese LN, Defour JP, Pecquet C, Constantinescu SN. The Thrombopoietin Receptor: Structural Basis of Traffic and Activation by Ligand, Mutations, Agonists, and Mutated Calreticulin. Frontiers in endocrinology. 2017:8():59. doi: 10.3389/fendo.2017.00059. Epub 2017 Mar 31 [PubMed PMID: 28408900]
Pecci A, Ragab I, Bozzi V, De Rocco D, Barozzi S, Giangregorio T, Ali H, Melazzini F, Sallam M, Alfano C, Pastore A, Balduini CL, Savoia A. Thrombopoietin mutation in congenital amegakaryocytic thrombocytopenia treatable with romiplostim. EMBO molecular medicine. 2018 Jan:10(1):63-75. doi: 10.15252/emmm.201708168. Epub [PubMed PMID: 29191945]
Germeshausen M, Ancliff P, Estrada J, Metzler M, Ponstingl E, Rütschle H, Schwabe D, Scott RH, Unal S, Wawer A, Zeller B, Ballmaier M. MECOM-associated syndrome: a heterogeneous inherited bone marrow failure syndrome with amegakaryocytic thrombocytopenia. Blood advances. 2018 Mar 27:2(6):586-596. doi: 10.1182/bloodadvances.2018016501. Epub [PubMed PMID: 29540340]
Level 3 (low-level) evidenceAmmeti D, Marzollo A, Gabelli M, Zanchetta ME, Tretti-Parenzan C, Bottega R, Capaci V, Biffi A, Savoia A, Bresolin S, Faleschini M. A novel mutation in MECOM affects MPL regulation in vitro and results in thrombocytopenia and bone marrow failure. British journal of haematology. 2023 Dec:203(5):852-859. doi: 10.1111/bjh.19023. Epub 2023 Aug 23 [PubMed PMID: 37610030]
Azuno Y, Yaga K. Successful cyclosporin A therapy for acquired amegakaryocytic thrombocytopenic purpura. American journal of hematology. 2002 Apr:69(4):298-9 [PubMed PMID: 11921030]
Level 3 (low-level) evidenceMulroy E, Gleeson S, Chiruka S. Danazol: an effective option in acquired amegakaryocytic thrombocytopaenic purpura. Case reports in hematology. 2015:2015():171253. doi: 10.1155/2015/171253. Epub 2015 Apr 5 [PubMed PMID: 25945269]
Level 3 (low-level) evidenceEvans DI. Immune amegakaryocytic thrombocytopenia of the newborn: association with anti-HLA-A2. Journal of clinical pathology. 1987 Mar:40(3):258-61 [PubMed PMID: 3494047]
Level 3 (low-level) evidenceKhan F, Shehna A, Mukundan L. Mononeuritis multiplex in acquired amegakaryocytic thrombocytopenia. Journal of neurosciences in rural practice. 2015 Oct-Dec:6(4):588-90. doi: 10.4103/0976-3147.165351. Epub [PubMed PMID: 26752909]
Ichikawa T, Shimojima Y, Otuki T, Ueno KI, Kishida D, Sekijima Y. Acquired Amegakaryocytic Thrombocytopenia in Adult-onset Still's Disease: Successful Combination Therapy with Tocilizumab and Cyclosporine. Internal medicine (Tokyo, Japan). 2019 Dec 1:58(23):3473-3478. doi: 10.2169/internalmedicine.2929-19. Epub 2019 Aug 6 [PubMed PMID: 31391399]
Manoharan A, Williams NT, Sparrow R. Acquired amegakaryocytic thrombocytopenia: report of a case and review of literature. The Quarterly journal of medicine. 1989 Mar:70(263):243-52 [PubMed PMID: 2690174]
Level 3 (low-level) evidenceDahal S, Sharma E, Dahal S, Shrestha B, Bhattarai B. Acquired Amegakaryocytic Thrombocytopenia and Pure Red Cell Aplasia in Thymoma. Case reports in hematology. 2018:2018():5034741. doi: 10.1155/2018/5034741. Epub 2018 Mar 11 [PubMed PMID: 29713553]
Level 3 (low-level) evidenceIkeda N, Hisano Y, Kamao T, Uno M, Mizushima T. Acquired Amegakaryocytic Thrombocytopenia Associated With Autoimmune Hemolytic Anemia. Cureus. 2022 Jul:14(7):e27315. doi: 10.7759/cureus.27315. Epub 2022 Jul 26 [PubMed PMID: 36042987]
Agarwal N, Spahr JE, Werner TL, Newton DL, Rodgers GM. Acquired amegakaryocytic thrombocytopenic purpura. American journal of hematology. 2006 Feb:81(2):132-5 [PubMed PMID: 16432869]
Level 3 (low-level) evidenceErgas D, Tsimanis A, Shtalrid M, Duskin C, Berrebi A. T-gamma large granular lymphocyte leukemia associated with amegakaryocytic thrombocytopenic purpura, Sjögren's syndrome, and polyglandular autoimmune syndrome type II, with subsequent development of pure red cell aplasia. American journal of hematology. 2002 Feb:69(2):132-4 [PubMed PMID: 11835350]
Level 3 (low-level) evidenceSavoia A, Dufour C, Locatelli F, Noris P, Ambaglio C, Rosti V, Zecca M, Ferrari S, di Bari F, Corcione A, Di Stazio M, Seri M, Balduini CL. Congenital amegakaryocytic thrombocytopenia: clinical and biological consequences of five novel mutations. Haematologica. 2007 Sep:92(9):1186-93 [PubMed PMID: 17666371]
Novotný JP, Köhler B, Max R, Egerer G. Acquired Amegakaryocytic Thrombocytopenic Purpura Progressing into Aplastic Anemia. Prague medical report. 2017:118(4):147-155. doi: 10.14712/23362936.2017.16. Epub [PubMed PMID: 29324222]
Kaushansky K. Thrombopoietin. The New England journal of medicine. 1998 Sep 10:339(11):746-54 [PubMed PMID: 9731092]
Level 3 (low-level) evidenceKim AR, Sankaran VG. Thrombopoietin: tickling the HSC's fancy. EMBO molecular medicine. 2018 Jan:10(1):10-12. doi: 10.15252/emmm.201708450. Epub [PubMed PMID: 29191946]
Hirata S, Takayama N, Jono-Ohnishi R, Endo H, Nakamura S, Dohda T, Nishi M, Hamazaki Y, Ishii E, Kaneko S, Otsu M, Nakauchi H, Kunishima S, Eto K. Congenital amegakaryocytic thrombocytopenia iPS cells exhibit defective MPL-mediated signaling. The Journal of clinical investigation. 2013 Sep:123(9):3802-14. doi: 10.1172/JCI64721. Epub 2013 Aug 1 [PubMed PMID: 23908116]
Germeshausen M, Ballmaier M. CAMT-MPL: congenital amegakaryocytic thrombocytopenia caused by MPL mutations - heterogeneity of a monogenic disorder - a comprehensive analysis of 56 patients. Haematologica. 2021 Sep 1:106(9):2439-2448. doi: 10.3324/haematol.2020.257972. Epub 2021 Sep 1 [PubMed PMID: 32703794]
Nishino S, Kodaka T, Sawada Y, Goka T, Gotoh Y, Tsunemine H, Takahashi T. Marked rebound thrombocytosis in response to glucocorticoids in a patient with acquired amegakaryocytic thrombocytopenia. Journal of clinical and experimental hematopathology : JCEH. 2018 Dec 13:58(4):166-170. doi: 10.3960/jslrt.18016. Epub 2018 Nov 9 [PubMed PMID: 30416171]
Katsumata Y, Suzuki T, Kuwana M, Hattori Y, Akizuki S, Sugiura H, Matsuoka Y. Anti-c-Mpl (thrombopoietin receptor) autoantibody-induced amegakaryocytic thrombocytopenia in a patient with systemic sclerosis. Arthritis and rheumatism. 2003 Jun:48(6):1647-51 [PubMed PMID: 12794833]
Level 3 (low-level) evidenceIchimata S, Kobayashi M, Honda K, Shibata S, Matsumoto A, Kanno H. Acquired amegakaryocytic thrombocytopenia previously diagnosed as idiopathic thrombocytopenic purpura in a patient with hepatitis C virus infection. World journal of gastroenterology. 2017 Sep 21:23(35):6540-6545. doi: 10.3748/wjg.v23.i35.6540. Epub [PubMed PMID: 29085203]
Ihara K, Ishii E, Eguchi M, Takada H, Suminoe A, Good RA, Hara T. Identification of mutations in the c-mpl gene in congenital amegakaryocytic thrombocytopenia. Proceedings of the National Academy of Sciences of the United States of America. 1999 Mar 16:96(6):3132-6 [PubMed PMID: 10077649]
Level 3 (low-level) evidenceObi EI, Osegi N, Etu-Efeotor TP, Pughikumo OC, Toluhi H, Makinde O. Massive hemoperitoneum from hemorrhagic corpus luteum in a patient with acquired amegakaryocytic thrombocytopenic purpura. Clinical case reports. 2020 Apr:8(4):719-721. doi: 10.1002/ccr3.2767. Epub 2020 Mar 7 [PubMed PMID: 32274044]
Level 3 (low-level) evidenceBallmaier M, Holter W, Germeshausen M. Flow cytometric detection of MPL (CD110) as a diagnostic tool for differentiation of congenital thrombocytopenias. Haematologica. 2015 Sep:100(9):e341-4. doi: 10.3324/haematol.2015.125963. Epub 2015 Apr 24 [PubMed PMID: 25911549]
Oved JH, Shah YB, Venella K, Paessler ME, Olson TS. Non-myeloablative conditioning is sufficient to achieve complete donor myeloid chimerism following matched sibling donor bone marrow transplant for myeloproliferative leukemia virus oncogene (MPL) mutation-driven congenital amegakaryocytic thrombocytopenia: Case report. Frontiers in pediatrics. 2022:10():903872. doi: 10.3389/fped.2022.903872. Epub 2022 Jul 28 [PubMed PMID: 35967582]
Level 3 (low-level) evidenceCancio M, Hebert K, Kim S, Aljurf M, Olson T, Anderson E, Burroughs L, Vatsayan A, Myers K, Hashem H, Hanna R, Horn B, Prestidge T, Boelens JJ, Boulad F, Eapen M. Outcomes in Hematopoietic Stem Cell Transplantation for Congenital Amegakaryocytic Thrombocytopenia. Transplantation and cellular therapy. 2022 Feb:28(2):101.e1-101.e6. doi: 10.1016/j.jtct.2021.10.009. Epub 2021 Oct 17 [PubMed PMID: 34670170]
Dragani M, Andreani G, Familiari U, Marci V, Rege-Cambrin G. Pure red cell aplasia and amegakaryocytic thrombocytopenia in thymoma: The uncharted territory. Clinical case reports. 2020 Apr:8(4):598-601. doi: 10.1002/ccr3.2642. Epub 2020 Mar 17 [PubMed PMID: 32274018]
Level 3 (low-level) evidenceKatai M, Aizawa T, Ohara N, Hiramatsu K, Hashizume K, Yamada T, Kitano K, Saito H, Shinoda T, Wakata S. Acquired amegakaryocytic thrombocytopenic purpura with humoral inhibitory factor for megakaryocyte colony formation. Internal medicine (Tokyo, Japan). 1994 Mar:33(3):147-9 [PubMed PMID: 8061390]
Level 3 (low-level) evidenceHussain SA, Fatima H, Faisal H, Bansal M. Acquired Amegakaryocytic Thrombocytopenia Progressing to Aplastic Anaemia. European journal of case reports in internal medicine. 2022:9(9):003479. doi: 10.12890/2022_003479. Epub 2022 Sep 5 [PubMed PMID: 36299833]
Level 3 (low-level) evidenceGupta L, Gupta V, Pani KC, Mittal N, Agarwal V. Successful use of azathioprine in glucocorticoid refractory immune amegakaryocytic thrombocytopenia of lupus. Reumatologia clinica. 2020 May-Jun:16(3):249-250. doi: 10.1016/j.reuma.2018.03.005. Epub 2018 Apr 17 [PubMed PMID: 29678667]
Tu X, Xue A, Wu S, Jin M, Zhao P, Zhang H. Case Report: Successful Avatrombopag Treatment for Two Cases of Anti-PD-1 Antibody-Induced Acquired Amegakaryocytic Thrombocytopenia. Frontiers in pharmacology. 2021:12():795884. doi: 10.3389/fphar.2021.795884. Epub 2022 Jan 27 [PubMed PMID: 35153753]
Level 3 (low-level) evidenceXue Y, Wu Q, Pu D, Xu F, Li Y. Radiation‑induced pure red cell aplasia combined with acquired amegakaryocytic thrombocytopenia in a thymoma after rapid response to radiotherapy: A case report and literature review. Experimental and therapeutic medicine. 2023 May:25(5):237. doi: 10.3892/etm.2023.11936. Epub 2023 Apr 4 [PubMed PMID: 37114181]
Level 3 (low-level) evidenceCleyrat C, Girard R, Choi EH, Jeziorski É, Lavabre-Bertrand T, Hermouet S, Carillo S, Wilson BS. Gene editing rescue of a novel MPL mutant associated with congenital amegakaryocytic thrombocytopenia. Blood advances. 2017 Sep 26:1(21):1815-1826. doi: 10.1182/bloodadvances.2016002915. Epub 2017 Sep 22 [PubMed PMID: 29296828]
Level 3 (low-level) evidenceFox NE, Lim J, Chen R, Geddis AE. F104S c-Mpl responds to a transmembrane domain-binding thrombopoietin receptor agonist: proof of concept that selected receptor mutations in congenital amegakaryocytic thrombocytopenia can be stimulated with alternative thrombopoietic agents. Experimental hematology. 2010 May:38(5):384-91. doi: 10.1016/j.exphem.2010.02.007. Epub 2010 Feb 24 [PubMed PMID: 20188141]
Level 3 (low-level) evidence