 Congenital Hypertrophy of Retinal Pigment Epithelium
Congenital Hypertrophy of Retinal Pigment Epithelium
Introduction
Congenital hypertrophy of the retinal pigmented epithelium (CHRPE) is a benign ocular condition characterized by well-defined, flat, pigmented lesions primarily located in the fundus of the eye.[1] CHRPE lesions are typically unilateral and solitary, although multiple or bilateral occurrences have been reported.[2] These lesions are usually discovered incidentally during routine ophthalmic examinations, often in asymptomatic individuals.[2] Fundoscopic examination may reveal solitary CHRPE lesions, which are more common, grouped lesions, or atypical multiple CHRPE lesions, often associated with disorders such as familial adenomatous polyposis (FAP) syndrome, Gardner syndrome, and Turcot syndrome.[3][4]
While generally considered benign, CHRPE lesions may occasionally undergo malignant transformation.[5][6] Thus, patients diagnosed with CHRPE should undergo a thorough systemic evaluation to exclude associated syndromes.[7][8] The management of isolated CHRPE lesions typically involves regular monitoring with ophthalmic examinations to detect any changes suggestive of malignant transformation or growth.[9] In cases where CHRPE lesions are associated with FAP syndrome, a multidisciplinary approach involving collaboration with gastroenterologists for colorectal cancer screening and genetic counseling is warranted.[10][11][12] While CHRPE lesions do not usually require treatment, patient education regarding the potential risk of malignant transformation and regular follow-up is essential.
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
CHRPE is a term commonly used to describe conditions that exhibit similar clinical features but vary significantly in etiology, prognosis, and management. Various studies have proposed the CHRPE names and abbreviations below to accurately describe and differentiate between these conditions.
Typical or solitary: This type of CHRPE is generally characterized by unilateral and solitary lesions not associated with systemic conditions. These lesions develop sporadically and are almost always benign. Typical CHRPEs are often regarded as the only true CHRPEs.[13]
Grouped or multifocal: These lesions are usually unilateral and appear similar to CHRPE but are multifocal. Grouped CHRPE lesions are clustered within a quadrant of the retina, often referred to as "bear tracks." They are congenital and benign, with no associated systemic conditions.[14]
Pigmented ocular fundus lesions of familial adenomatous polyposis: Also referred to as multiple CHRPEs or atypical CHRPEs, pigmented ocular fundus lesions of FAP (PO-FLs) are multifocal, similar to grouped CHRPEs.[15] However, these lesions are typically bilateral. Unlike other CHRPE variants, PO-FLs are associated with a group of hereditary genetic colonic polyposis conditions.[16] The location of the adenomatous polyposis coli (APC) gene mutation may influence the expression of PO-FLs.[17]
FAP is an autosomal dominant inherited condition resulting from a mutation in the APC tumor suppressor gene (chromosome 5q21–q22).[16] Severe FAP, characterized by more than 1000 colorectal adenomas, is associated with APC mutations between codons 1250 and 1464.[18] Attenuated FAP, with fewer than 100 colorectal adenomas, is attributed to mutations before codon 157, in the spliced region of exon 9, and mutations after 1595. All other mutations lead to intermediate FAP.[18][19] Mutations between codon 446 and codon 1338 are associated with multiple CHRPE lesions, while mutations between codons 1445 and 1578 are not.[20]
FAP is characterized by the development of hundreds of polyps within the colon that progress to colorectal cancer in nearly all untreated patients.[21][8] FAP often presents with extracolonic manifestations, including CHRPE-like lesions in the retina.[14] Gardner syndrome and Turcot syndrome are variants of FAP that are also associated with the development of PO-FLs.[22] Due to the potentially dire prognosis of this condition, eye care clinicians must be able to distinguish PO-FLs from their benign counterparts.[23]
Epidemiology
Prevalence rates of typical CHRPE have not been well defined in the literature and show significant variability between studies, ranging from 0.4% to 30%.[24] Coleman et al found a prevalence rate of 1.20% among a group of 1745 study subjects, subjects, with only 2 of these subjects presenting with multifocal lesions, accounting for a prevalence of less than 1%, and both lesions were unilateral in presentation.[13] Numerous studies have found no sexual preference for typical CHRPE lesions.[13]
Mirinezhad et al noted the presence of CHRPE in 78% of patients with FAP and 38% of their siblings.[25] Multiple CHRPEs may be found in up to 90% of patients with FAP.[26][8] FAP has a prevalence of approximately 1 in 8300 to 1 in 37,600 individuals.[27] On average, FAP occurs in 1 in 10,000 individuals.[21] In a systematic literature review, the mean specificity of CHRPE as a phenotypic marker of FAP was 89%, with a sensitivity of 79%.[8]
Pathophysiology
Specific histological changes in retinal pigment epithelial (RPE) cells and disruptions of retinal homeostasis underlie the development of retinal and CHRPE lesions.[1] Solitary and multifocal CHRPEs represent sporadic, benign abnormalities of RPE cells, also referred to as retinal hamartomas. Histopathological changes in RPE cell size, number, and content lead to cellular dysfunction and the loss of homeostasis among the RPE, choroid, and neural retina in the affected area.
Histopathology
Although CHRPE refers to specific hypertrophic cellular changes, histological analysis shows various morphologic cellular changes of CHRPE and its variants. RPE cells in typical CHRPEs exhibit various degrees of cellular hypertrophy. In hypertrophy, normal, cuboidal RPE cells are vertically elongated and columnar. Moreover, multiple studies have indicated that cells also undergo hyperplasia, leading to thickening of the RPE layer. Large melanosomes or macromelanosomes are evident in the RPE.[28] These round pigment granules contrast with the wedge-shaped melanin granules in normal RPE. These changes disrupt the ability of the RPE to support the overlying photoreceptors, resulting in cell loss and thinning.[29]
In the absence of photoreceptors, the production of lipofuscin, a waste product secondary to the normal RPE-mediated phagocytosis of photoreceptor outer segments, is diminished.[14] Atrophic areas, known as lacunae, may also develop throughout the lesion and correlate with a depigmented funduscopic appearance. Compared to normal cuboidal RPE cells, affected RPE cells are vertically elongated and range from cuboidal to columnar. They possess large, spherical intracytoplasmic melanin granules of much higher density than normal RPE cells. In addition, the adjacent Bruch membrane is thickened due to the thickening of the basement membrane of the RPE.[30] The choroid, choriocapillaris, and other layers of the Bruch membrane are normal.
Conversely, grouped CHRPE has been observed to contain histologically normal cells, showing no variance in cell size or density compared to normal RPE cells. Instead, its hyperpigmented appearance results from the increased density of melanin granules, which are large and ellipsoid in shape. Bruch membrane is normal in these cases.[31] Several atypical CHRPEs exhibit histological similarities to typical lesions, featuring hypertrophic and hyperplastic RPE cells along with thickening of the adjacent Bruch membrane.[13][14] In addition, retinal vascular changes or retinal invasion may be observed.
In FAP (ie, Gardner syndrome), a generalized abnormality of melanogenesis in the RPE may be observed. The lesions associated with FAP may manifest in at least 3 distinct appearances.[32] The first pattern is similar to typical solitary CHRPE. The second pattern resembles an RPE hamartoma with RPE hyperplasia and hypertrophy.[32] Minute mushroom-shaped lesions characterize the third pattern.[32] Some chronic lesions may penetrate the full thickness of the retina, thus classifying lesions associated with FAP as multiple RPE hamartomas related to FAP or polyposis-associated congenital hamartoma of the RPE.[1]
History and Physical
Solitary or Unifocal Congenital Hypertrophy of the Retinal Pigmented Epithelium
Solitary CHRPEs are typically flat (98.5%) and highly pigmented, appearing dark gray or black. However, approximately 1.5% of CHRPEs do not appear flat due to the presence of an intralesional nodule, which may increase with time (see Image. Congenital Hypertrophy of the Retinal Pigmented Epithelium [CHRPE]).[2] The color is consistent across different racial backgrounds. In some cases, lesions may become entirely depigmented (12%).[2] Usually, these lesions are round with well-defined smooth or scalloped borders. In about 52% of cases, a hypopigmented ring or "halo" surrounds the lesion. As the lesion matures, depigmented punched-out lacunae and a halo may develop, potentially involving the entire lesion (see Image. CHRPE With Lacunae).
Solitary CHRPEs are predominantly found in the equatorial fundus (45%), with the inferotemporal fundus being the most common location (31%).[2] In rare cases, lesions may be peripapillary (1%) or macular in location (1%).[33][2] Foveal involvement or choroidal neovascularization may lead to reduced visual acuity.[34][35] In a large study on 330 patients with solitary CHRPE, the median age of presentation was 45, with an age range between 1 and 80.[2] This study found no association between solitary CHRPE and FAP or colon cancer, although 8% of patients had a history of cancer, with breast cancer being the most common.[2][33] Approximately half (43%) of pigmented CHRPEs present with hypopigmented areas of RPE atrophy, known as lacunae, within the lesion.[2] On average, about 56.5% of the lesion surface is atrophic; however, the atrophy's presence, appearance, and extent vary between patients and do not change the patient's prognosis.
The mean largest basal diameter of solitary CHRPE is approximately 4.7 mm (median 4.5 mm).[2] However, lesions can vary in size, ranging from approximately 100 µm (smaller) to occupying the entire ocular fundus quadrant (larger). Although lesions are generally regarded as nonprogressive and stationary, numerous studies have demonstrated slow, benign flat enlargement over time in up to 83% of patients, for which the percentage of area occupied by lacunae within the CHRPE is an important factor.[36][2] The median rate of flat enlargement of solitary CHRPE is around 10 µm/month.[2] In addition, 32% of cases also show enlargement of lacunae in size or number over time.[2] Intraretinal focal pigmentation may be noted near the margin of solitary CHRPE. Lesions are often found incidentally during a routine eye examination, and patients are rarely symptomatic. Visual field defects corresponding to the area of the lesion are possible; however, lesions are generally located peripherally and are unlikely to be noticeable.
Grouped or Multifocal Congenital Hypertrophy of the Retinal Pigmented Epithelium and Grouped Pigmentation of the Retina
Grouped pigmentation of the retina (GPR) resembles animal footprints or bear tracks. GPR appears similar to solitary CHRPE but presents unilaterally with 3 to 30 discreet lesions of various sizes (see Image. Bear Track Pigmented Lesions). These lesions are typically flat, round, or oval black spots with well-defined margins arranged in clusters. GPR lesions are generally smaller than unilateral CHRPEs, ranging from 0.1 to 0.3 mm in diameter, and confined to a single quadrant in the retina. Peripheral lesions are usually larger than posterior lesions and seem to radiate from the optic disc along the lines of Blashko lines or to proceed toward the retinal periphery.[37] Unlike solitary CHRPE, GPR lesions typically lack surrounding haloes and lacunae.[1]
Usually, several groups or clusters of such lesions are present, mostly affecting an eye. Rarely, such lesions may appear depigmented, congenital grouped albinotic spots, also called "polar bear tracks." These grouped CHRPE lesions are also not associated with gastrointestinal malignancy. Meyer et al reported a male patient with bear tracks in the left eye and pigmentation in the left arm along Blashko lines.[38] They hypothesized that the mosaic pigmentation of skin and eye on the same side may be related to the common etiology of early postzygotic mutation.[38] The pigmentation pattern of GPR does not align with the retinal nerve fiber layer, suggesting RPE cell outgrowth and migration during embryogenesis.[37]
Pigmented Ocular Fundus Lesions of Familial Adenomatous Polyposis and Familial Adenomatous Polyposis–Associated Lesions
PO-FLs, also called multiple CHRPEs or atypical CHRPEs, must be accurately differentiated from benign CHRPEs due to their association with severe systemic disease. Compared to their benign counterpart, PO-FLs are characterized by their bilateral and multifocal nature, extending across multiple quadrants. They commonly exhibit a "pisciform" (fish-shaped) appearance with irregular, depigmented borders (see Image. CHRPE Associated With FAP).[14] These lesions may vary in shape, including spindle, oval, comma, or fishtail, and can display different pigmentation patterns such as light gray, brown, or black. The size of PO-FLs may vary in the same eye but is usually smaller, approximately 50 to 100 µm, than solitary CHRPEs. They often exhibit irregularly depigmented or hypopigmented margins and surrounding areas. Notably, features such as retinal invasion, proliferation of RPE, glial cells, and capillaries, as well as depigmented halo, lacunae, RPE changes near retinal vessels, mottling, and pigmented satellite lesions may be present. These lesions are typically distributed in a haphazard pattern across the retina rather than in a sectorial arrangement.
PO-FLs are congenital and have also been reported in preterm infants.[39] The presence of more than 4 widely spaced multiple CHRPEs in both eyes may suggest FAP or Gardner syndrome.[40] However, the absence of atypical CHRPEs does not rule out FAP. Bonnet et al suggested specific criteria with high specificity and positive predictive value for detecting FAP in PO-FLs, including:
- Multiple lesions with a total of at least 3 lesions in 1 or both eyes
- Lesion type A or B according to Berk classification [41]
- Bilaterality [8]
The Berk classification of CHRPEs categorizes them into the following 4 types:
Evaluation
Dilated Fundus Examination
Detecting CHRPE lesions often requires evaluating the retinal periphery through dilated examination or nonmydriatic retinal imaging, as they are rarely found in the posterior pole. One or both procedures are virtually always performed as part of a comprehensive eye exam. Most biomicroscopes are equipped with a red-free filter that may aid in distinguishing between choroidal nevi and CHRPE lesions. Nevi disappears with red-free light, whereas CHRPE remains visible.[42]
Optical Coherence Tomography
Optical coherence tomography (OCT) reveals hyperreflectivity and thickening of the RPE, corresponding to elongated RPE cells. This thickening causes shadowing of the underlying choroid, with resultant retinal thinning and photoreceptor layer loss over the lesion.[43] Francis et al found evidence of associated choroidal atrophy when lesions were imaged with spectral domain OCT (SD-OCT). They found that the inner layers of the choroid (ie, the choriocapillaris and Sattler layer) were atrophied in 97% of their cohort. Larger lesions were more likely to involve the outer choroid (Haller's layer), as noted in 53% of patients.[44] Lacunae exhibit RPE loss and increased light transmission, potentially affecting visual function. Visual field loss or microperimetry changes may be attributed to photoreceptor atrophy, with subretinal clefts observed in certain cases.
Fluorescein Angiography
Fluorescein angiography reveals hypofluorescence of lesions due to the "block effect," caused by hypertrophic RPE cells anterior to the choroidal vasculature, obstructing the visualization of normal background fluorescein leakage. If present, lacunae within lesions will appear hyperfluorescent.[45] In some cases, changes in retinal vasculature may occur, which can be detected with fluorescein angiography, including capillary nonperfusion areas, neovascularization, capillary microaneurysms, dye leakage, and chorioretinal anastomosis. Polar bear tracks show early and late hyperfluorescence consistent with window defects.[46]
Indocyanine Green Angiogram
Indocyanine green angiography similarly depicts blocked fluorescence within the region of pigmented lesions, although choroidal vessels may be visible through the lacunae.
Fundus Autofluorescence
Fundus autofluorescence imaging typically reveals lesions as strongly hypoautofluorescent, although lacunae may exhibit slight hyperautofluorescence due to their melanin-rich composition and limited presence of lipofuscin.[47] Polar bear tracks also show hypoautofluorescence, possibly due to RPE atrophy.[46] An alternate theory suggests the absorption or blockage of normal autofluorescence by abnormal melanin precursor material.[46]
B-Scan Ultrasonography
B-scan ultrasonography is not typically diagnostic for CHRPE but can assist in measuring the thickness of the lesion. Ultrasound A or B scan may not detect or document these lesions, as these lesions are flat.[1]
Visual Field Analysis
Although patients can manifest a visual field defect corresponding to the area of the CHRPE, they are unlikely to be detected through visual field analysis. This is because most lesions are situated more peripherally than the testing area covered by Humphrey visual field analyzers. The depth of the defect (absolute versus relative) usually correlates with lesion size. However, some studies suggest that younger patients typically manifest relative visual field defects, while older patients manifest absolute defects.[43]
Optical Coherence Tomography AngiogramIn solitary CHRPE cases, OCT angiography typically shows normal retinal vasculature, although interpretation can be challenging due to segmentation artifacts caused by retinal thinning.[48] The pigmented lesion largely blocks the choroidal vasculature. En-face OCT provides a clearer analysis of the normal choroidal vasculature in these cases.[48]
Electrophysiology
In eyes with CHRPE, electroretinography and electro-oculography typically yield normal results.[49]
Systemic Evaluation
Systemic evaluation and genetic counseling should be conducted in patients with atypical multiple CHRPEs to exclude mutations in the APC gene and APC-associated polyposis conditions, including FAP, Gardner syndrome, and Turcot syndrome.[22]
FAP is an autosomal dominant disorder caused by a germline mutation in the APC gene, typically leading to the development of colon carcinoma in patients by age 50. Adenomatous polyps in the colon and rectum usually emerge during adolescence, necessitating regular colonoscopies with or without polyp excision. Atypical multiple CHRPE lesions are prevalent in 70% to 90% of FAP patients.[13] Some atypical CHRPE cases manifest lesions from birth, highlighting the importance of meticulous screening, genetic testing, and regular examination for all family members due to autosomal dominant inheritance.
Gardner syndrome is a condition characterized by FAP along with soft tissue tumors, including lipoma, fibroma, and epidermoid cysts, as well as osteomas of the mandible, skull, and long bone. Turcot syndrome, an autosomal recessive condition, presents with brain tumors such as medulloblastoma and gliomas in association with FAP.
Treatment / Management
Solitary CHRPE and GPR do not require treatment and can be monitored periodically. Solitary lesions should undergo close examination to detect rare nodule development and associated complications or malignant transformation (Please refer to the Prognosis section for more information). The lesion's dimensions can be measured, and fundus photography can be utilized to monitor changes in size and characteristics.
Patients with PO-FLs should undergo routine endoscopies with their gastroenterologist to assess the presence of polyps within the colon and rectum. Genetic screening should be conducted in patients with a family history of adenomatous polyposis syndrome or patients with 10 or more polyps, regardless of family history.[50] The specific mutation identified through genetic testing guides the recommended frequency of screening and prognosis. However, solitary or multifocal CHRPEs are not associated with an increased risk of systemic disease and do not require further testing or referral.[14](B3)
Differential Diagnosis
Differential diagnoses of CHRPE include:
- Choroidal nevus: Margins are not well defined, lesions lack the deep black appearance of CHRPE, and lacunae are absent. Overlying drusen may be observed.
- Choroidal melanoma: Lesions are often elevated, with margins less sharp compared to CHRPE.
- Melanocytoma of the optic nerve head: This condition typically presents with a feathery and elevated appearance.
- Congenital simple RPE hamartoma: Usually located in the posterior pole, with the margin often discernible with the lesion at the retina's inner surface on OCT.[51]
- Combined hamartoma of the retina and RPE: Typically observed in young patients with an epiretinal membrane or vitreomacular traction and pigmentary changes. OCT macula may exhibit a characteristic "omega sign."[52][53]
- Acquired RPE hyperplasia.[13]
- Focal pigmentation.
- Black sunburst related to sickle cell retinopathy.
- Healed choroiditis or chorioretinitis.[54]
- Torpedo maculopathy: Characterized by a torpedo-like appearance, typically located temporal to the fovea.[55]
- Paving stone degeneration: Usually present in the extreme retinal periphery near the ora, commonly as multiple lesions noted in inferior or inferotemporal areas.
Prognosis
Virtually all patients with solitary CHRPE or GPR are asymptomatic, and the lesions are benign. They are not associated with an increased risk of systemic disease. The prognosis is excellent. However, PO-FLs are associated with FAP, which can be fatal due to colon cancer. Therefore, regular ocular follow-ups and colonoscopies are necessary for prompt detection and management of colon polyps or cancer.
Complications
Complications associated with CHRPE are exceedingly rare and are generally secondary to nodular growth within the lesion. Subretinal fluid or exudation from nodules may lead to retinal detachment, characterized by the separation of the RPE from the neural retina. In addition, nodules may cause secondary vitreomacular traction and decreased central vision.[1] Although rare (2%), evidence of benign (adenoma) or malignant (adenocarcinoma) nodular transformation of RPE cells within solitary CHRPE lesions has been reported.[56][36] These transformations typically originate from the CHRPE as small pigmented nodules that grow slowly, eventually invading the sensory retina and generating their vasculature.
Feeder and drainage vessels carry lipoproteinaceous material to and from the lesion. Nodules may remain stable or progress to a large pedunculated lesion, potentially causing extensive intraocular inflammation, subretinal exudation, retinal detachment, and severe vision loss.[36] Consequently, long-term follow-up of all CHRPE lesions is necessary. Exudative maculopathy associated with presumed RPE adenoma related to CHRPE may respond to multiple intravitreal anti-vascular endothelial growth factor agents, such as bevacizumab.[57] Proton beam radiotherapy represents another option for managing RPE adenocarcinoma arising from CHRPE.[58] Additional complications include vitreous traction, cataracts, and posterior synechiae. However, uncomplicated cases are often monitored, as incidences of metastasis have not been reported.[36] Solitary CHRPE is not associated with an increased risk of gastrointestinal malignancy.
Deterrence and Patient Education
Patients should receive education about the diagnosis of CHRPE, with a focus on the significance of regular eye examinations. They should also be informed about the possibility of transformation and reassured that malignancy is exceedingly rare and unlikely in cases of solitary CHRPE.[1] Upon initial presentation of suspected PO-FLs, patients should undergo thorough education about the association with systemic polyposis syndromes and the increased risk of colon cancer development.
Patients should be advised to undergo further evaluation by a gastroenterologist and to adhere to regular screening protocols, as almost all patients with FAP eventually develop colon cancer.[14][11] Patients with FAP should undergo sigmoidoscopy or colonoscopy every 1 to 2 years, starting from age 10 to 12.[11] In addition, patients should schedule colonoscopies when polyps are detected.[11] Although multiple CHRPEs or PO-FLs are observed in a significant number of FAP cases, management should include eye examinations, genetic testing, and colonoscopy.[7][14]
Enhancing Healthcare Team Outcomes
In the context of CHRPE, a comprehensive approach involving various healthcare professionals is crucial to optimize patient-centered care, outcomes, patient safety, and team performance. Eye care professionals, mainly optometry and ophthalmology clinicians, are responsible for the initial identification of solitary CHRPE and GPR. However, interprofessional collaboration is essential due to the complexity of suspicious cases and potential systemic implications. This collaboration involves physicians, advanced practitioners, nurses, pharmacists, and other specialists.
An interprofessional approach is essential for managing PO-FLs, given their association with severe systemic conditions. This strategy involves primary care clinicians, nurses, pharmacists, geneticists, oncologists, plastic surgeons, orthopedists, and gastroenterologists. Each healthcare team member has specific responsibilities, such as providing expertise, facilitating communication, ensuring timely referrals, and coordinating care. Effective communication channels are vital for sharing information, discussing treatment options, and addressing patient concerns. Through collaborative efforts, healthcare professionals can ensure comprehensive evaluation, accurate diagnosis, and optimal management of CHRPE, leading to improved patient outcomes, safety, and overall team performance.
Media
(Click Image to Enlarge)
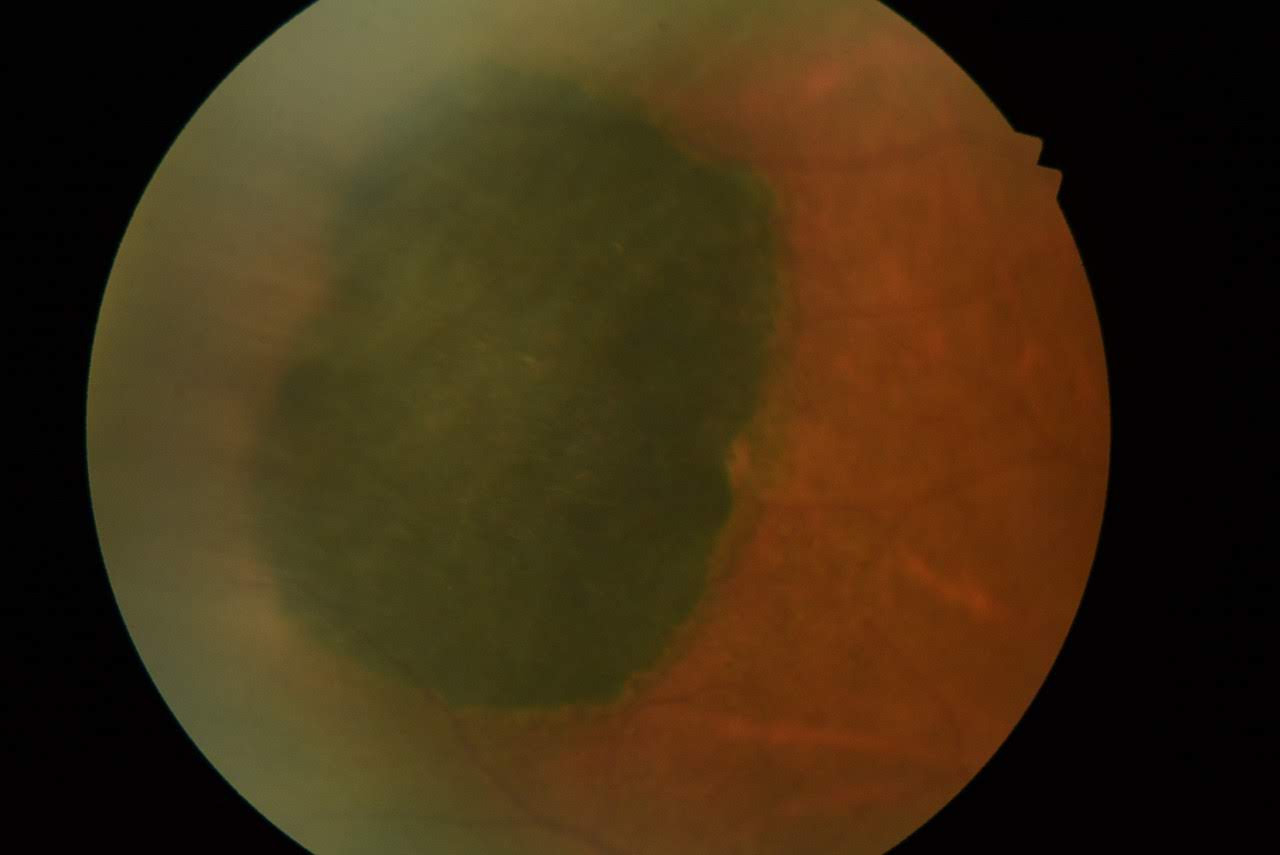
Congenital Hypertrophy of the Retinal Pigmented Epithelium (CHRPE). Solitary CHRPE lesions are typically flat and highly pigmented, and they present as dark gray or black areas.
Contributed by K Tripathy, MD
(Click Image to Enlarge)
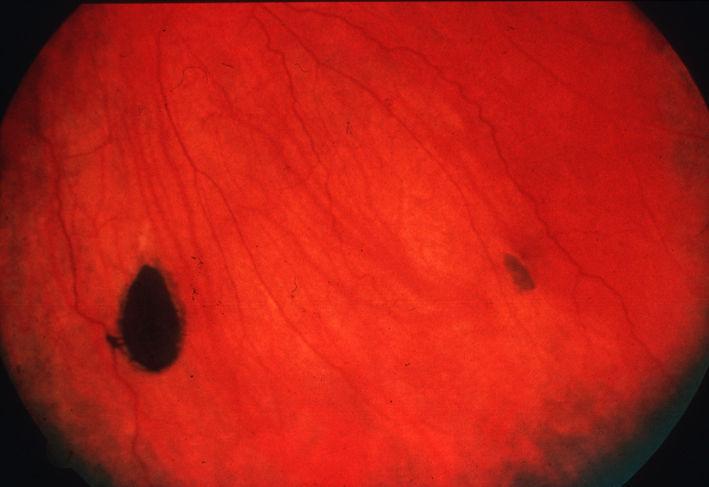
CHRPE With Lacunae. This image depicts a case of congenital hypertrophy of the retinal pigmented epithelium (CHRPE) with depigmented punched-out lacunae and a depigmented halo around the solitary lesion. These features become more prominent as the lesion matures and may eventually involve the entire lesion.
Contributed by K Tripathy, MD
(Click Image to Enlarge)
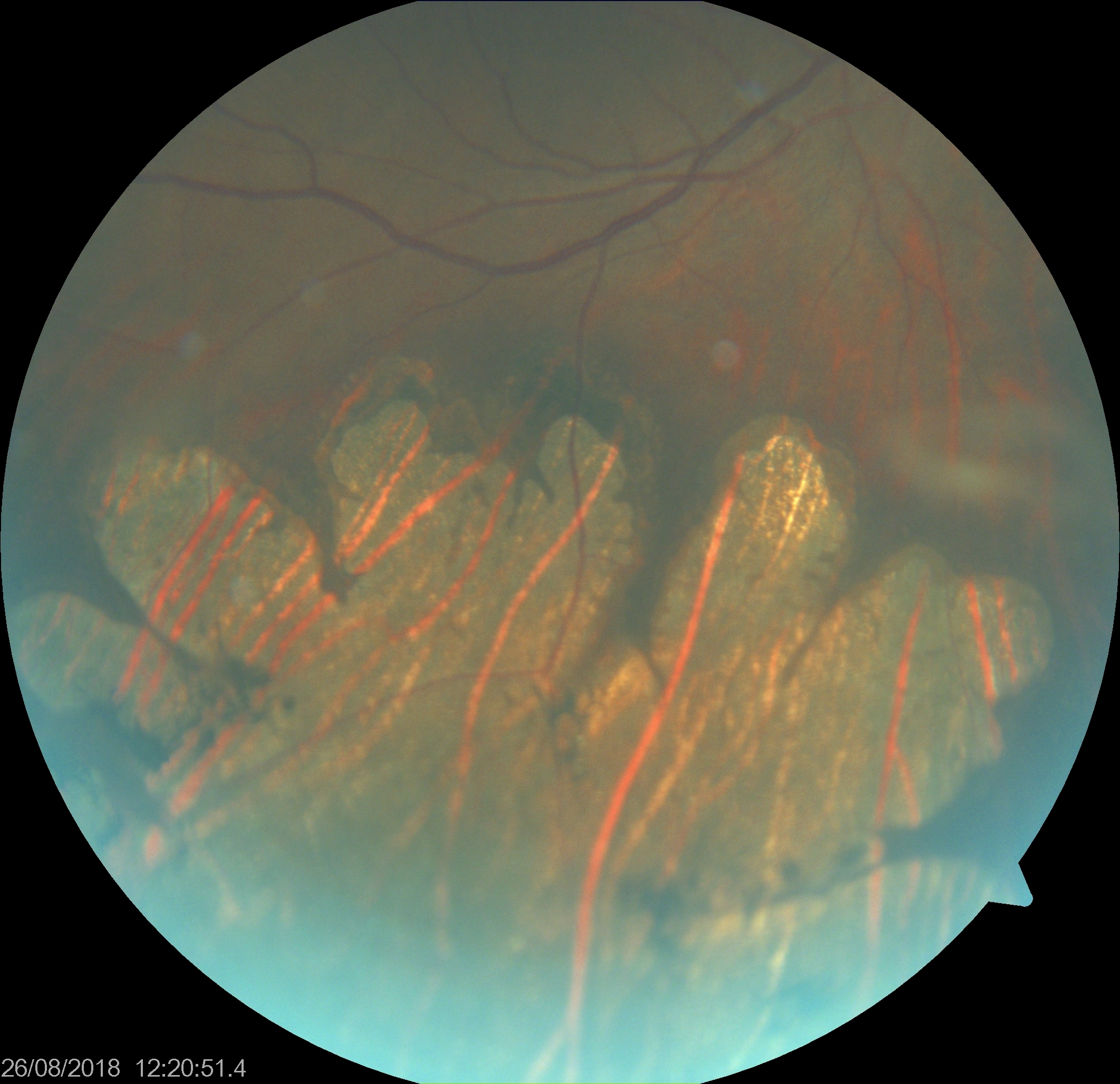
Bear Track Pigmented Lesions. Grouped pigmentation of the retina (GPR) resembles animal footprints or bear tracks. While similar to solitary congenital hypertrophy of the retinal pigmented epithelium, GPR presents unilaterally with approximately 3 to 30 discreet lesions of varying sizes. These lesions are typically flat, round, or oval black spots with well-defined margins arranged in clusters.
Contributed by K Tripathy, MD
(Click Image to Enlarge)
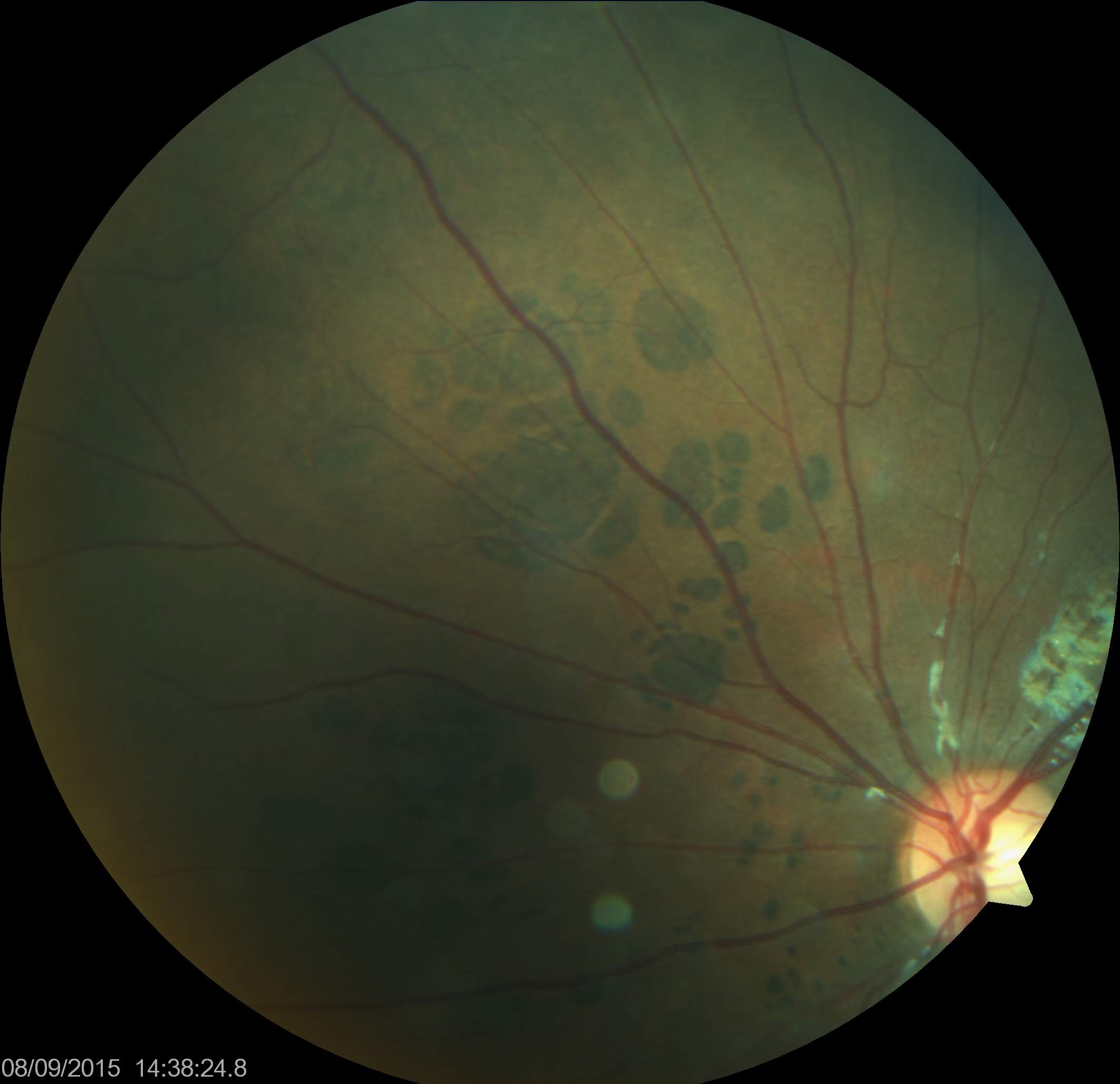
CHRPE Associated With FAP. Congenital hypertrophy of the retinal pigmented epithelium (CHRPE) associated with familial adenomatous polyposis (FAP) often exhibits a "pisciform" (fish-shaped) appearance with irregular, depigmented borders.
Filip em, Public Domain, via Wikimedia Commons
References
Shields JA, Shields CL. Tumors and Related Lesions of the Pigmented Epithelium. Asia-Pacific journal of ophthalmology (Philadelphia, Pa.). 2017 Mar-Apr:6(2):215-223. doi: 10.22608/APO.201705. Epub [PubMed PMID: 28399346]
Shields CL, Mashayekhi A, Ho T, Cater J, Shields JA. Solitary congenital hypertrophy of the retinal pigment epithelium: clinical features and frequency of enlargement in 330 patients. Ophthalmology. 2003 Oct:110(10):1968-76 [PubMed PMID: 14522773]
Liu Y, Moore AT. Congenital focal abnormalities of the retina and retinal pigment epithelium. Eye (London, England). 2020 Nov:34(11):1973-1988. doi: 10.1038/s41433-020-0902-4. Epub 2020 May 4 [PubMed PMID: 32367006]
Tiret A, Taiel-Sartral M, Tiret E, Laroche L. Diagnostic value of fundus examination in familial adenomatous polyposis. The British journal of ophthalmology. 1997 Sep:81(9):755-8 [PubMed PMID: 9422927]
Shields JA, Shields CL, Eagle RC Jr, Singh AD. Adenocarcinoma arising from congenital hypertrophy of retinal pigment epithelium. Archives of ophthalmology (Chicago, Ill. : 1960). 2001 Apr:119(4):597-602 [PubMed PMID: 11296028]
Sreenivasan J, Rishi P, Das K, Krishnakumar S, Biswas J. Retinal Pigment Epithelium Adenoma and Adenocarcinoma: A Review. Ocular oncology and pathology. 2021 Mar:7(2):121-132. doi: 10.1159/000509484. Epub 2020 Dec 22 [PubMed PMID: 33981695]
Rehan S, Aye K. In patients with a positive family history of familial adenomatous polyposis can the condition be diagnosed from the presence of congenital hypertrophy of the retinal pigment epithelium detected via an eye examination: A systematic review. Clinical & experimental ophthalmology. 2020 Jan:48(1):98-116. doi: 10.1111/ceo.13643. Epub 2019 Oct 10 [PubMed PMID: 31525261]
Level 1 (high-level) evidenceBonnet LA, Conway RM, Lim LA. Congenital Hypertrophy of the Retinal Pigment Epithelium (CHRPE) as a Screening Marker for Familial Adenomatous Polyposis (FAP): Systematic Literature Review and Screening Recommendations. Clinical ophthalmology (Auckland, N.Z.). 2022:16():765-774. doi: 10.2147/OPTH.S354761. Epub 2022 Mar 15 [PubMed PMID: 35321042]
Level 1 (high-level) evidenceBraga CS, Ricardo OMP, Cordeiro FM, Vieira JM, Nogueira FB. Suspect asymptomatic lesions: Congenital hypertrophy of the Retinal Pigment Epithelium (CHRPE). Romanian journal of ophthalmology. 2021 Jul-Sep:65(3):275-278. doi: 10.22336/rjo.2021.55. Epub [PubMed PMID: 35036651]
Hyer W, Cohen S, Attard T, Vila-Miravet V, Pienar C, Auth M, Septer S, Hawkins J, Durno C, Latchford A. Management of Familial Adenomatous Polyposis in Children and Adolescents: Position Paper From the ESPGHAN Polyposis Working Group. Journal of pediatric gastroenterology and nutrition. 2019 Mar:68(3):428-441. doi: 10.1097/MPG.0000000000002247. Epub [PubMed PMID: 30585891]
Yang J, Gurudu SR, Koptiuch C, Agrawal D, Buxbaum JL, Abbas Fehmi SM, Fishman DS, Khashab MA, Jamil LH, Jue TL, Law JK, Lee JK, Naveed M, Qumseya BJ, Sawhney MS, Thosani N, Wani SB, Samadder NJ. American Society for Gastrointestinal Endoscopy guideline on the role of endoscopy in familial adenomatous polyposis syndromes. Gastrointestinal endoscopy. 2020 May:91(5):963-982.e2. doi: 10.1016/j.gie.2020.01.028. Epub 2020 Mar 10 [PubMed PMID: 32169282]
Vasen HF, Möslein G, Alonso A, Aretz S, Bernstein I, Bertario L, Blanco I, Bülow S, Burn J, Capella G, Colas C, Engel C, Frayling I, Friedl W, Hes FJ, Hodgson S, Järvinen H, Mecklin JP, Møller P, Myrhøi T, Nagengast FM, Parc Y, Phillips R, Clark SK, de Leon MP, Renkonen-Sinisalo L, Sampson JR, Stormorken A, Tejpar S, Thomas HJ, Wijnen J. Guidelines for the clinical management of familial adenomatous polyposis (FAP). Gut. 2008 May:57(5):704-13. doi: 10.1136/gut.2007.136127. Epub 2008 Jan 14 [PubMed PMID: 18194984]
Coleman P, Barnard NA. Congenital hypertrophy of the retinal pigment epithelium: prevalence and ocular features in the optometric population. Ophthalmic & physiological optics : the journal of the British College of Ophthalmic Opticians (Optometrists). 2007 Nov:27(6):547-55 [PubMed PMID: 17956359]
Level 2 (mid-level) evidenceDeibert B, Ferris L, Sanchez N, Weishaar P. The link between colon cancer and congenital hypertrophy of the retinal pigment epithelium (CHRPE). American journal of ophthalmology case reports. 2019 Sep:15():100524. doi: 10.1016/j.ajoc.2019.100524. Epub 2019 Jul 24 [PubMed PMID: 31384696]
Level 3 (low-level) evidenceLi CJ, Yaghy A, Shields CL. Pigmented Ocular Fundus Lesions Associated With Familial Adenomatous Polyposis. Ophthalmic surgery, lasers & imaging retina. 2020 Feb 1:51(2):124. doi: 10.3928/23258160-20200129-10. Epub [PubMed PMID: 32084287]
Traboulsi EI, Apostolides J, Giardiello FM, Krush AJ, Booker SV, Hamilton SR, Hussels IE. Pigmented ocular fundus lesions and APC mutations in familial adenomatous polyposis. Ophthalmic genetics. 1996 Dec:17(4):167-74 [PubMed PMID: 9010867]
Valanzano R, Cama A, Volpe R, Curia MC, Mencucci R, Palmirotta R, Battista P, Ficari F, Mariani-Costantini R, Tonelli F. Congenital hypertrophy of the retinal pigment epithelium in familial adenomatous polyposis. Novel criteria of assessment and correlations with constitutional adenomatous polyposis coli gene mutations. Cancer. 1996 Dec 1:78(11):2400-10 [PubMed PMID: 8941012]
Tanabe H, Ijiri M, Takahashi K, Sasagawa H, Kamanaka T, Kuroda S, Sato H, Sarashina T, Mizukami Y, Makita Y, Okumura T. Genomic insights into familial adenomatous polyposis: unraveling a rare case with whole APC gene deletion and intellectual disability. Human genome variation. 2024 Mar 29:11(1):13. doi: 10.1038/s41439-024-00270-3. Epub 2024 Mar 29 [PubMed PMID: 38548799]
Level 3 (low-level) evidenceNieuwenhuis MH, Vasen HF. Correlations between mutation site in APC and phenotype of familial adenomatous polyposis (FAP): a review of the literature. Critical reviews in oncology/hematology. 2007 Feb:61(2):153-61 [PubMed PMID: 17064931]
Caspari R, Olschwang S, Friedl W, Mandl M, Boisson C, Böker T, Augustin A, Kadmon M, Möslein G, Thomas G. Familial adenomatous polyposis: desmoid tumours and lack of ophthalmic lesions (CHRPE) associated with APC mutations beyond codon 1444. Human molecular genetics. 1995 Mar:4(3):337-40 [PubMed PMID: 7795585]
Menon G, Carr S, Kasi A. Familial Adenomatous Polyposis. StatPearls. 2024 Jan:(): [PubMed PMID: 30855821]
Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, Yen T, Stanich PP, Axell L, Patel SG. APC-Associated Polyposis Conditions. GeneReviews(®). 1993:(): [PubMed PMID: 20301519]
McKay DL. Congenital hypertrophy of the retinal pigment epithelium and familial adenomatous polyposis. Australian and New Zealand journal of ophthalmology. 1993 Feb:21(1):3-6 [PubMed PMID: 8388223]
Chen CS, Phillips KD, Grist S, Bennet G, Craig JE, Muecke JS, Suthers GK. Congenital hypertrophy of the retinal pigment epithelium (CHRPE) in familial colorectal cancer. Familial cancer. 2006:5(4):397-404 [PubMed PMID: 16944273]
Mirinezhad SK, Mousavi F, Baghri M, Sepehri B, Ghavidel A, Ghojazadeh M, Somi MH. Congenital Hypertrophy of Retinal Pigment Epithelium for Diagnosis of Familial Adenomatous Polyposis - the First FAP registry in Iran. Asian Pacific journal of cancer prevention : APJCP. 2018 Jan 27:19(1):167-169 [PubMed PMID: 29373909]
Nusliha A, Dalpatadu U, Amarasinghe B, Chandrasinghe PC, Deen KI. Congenital hypertrophy of retinal pigment epithelium (CHRPE) in patients with familial adenomatous polyposis (FAP); a polyposis registry experience. BMC research notes. 2014 Oct 18:7():734. doi: 10.1186/1756-0500-7-734. Epub 2014 Oct 18 [PubMed PMID: 25326340]
Half E, Bercovich D, Rozen P. Familial adenomatous polyposis. Orphanet journal of rare diseases. 2009 Oct 12:4():22. doi: 10.1186/1750-1172-4-22. Epub 2009 Oct 12 [PubMed PMID: 19822006]
Level 3 (low-level) evidenceLloyd WC 3rd, Eagle RC Jr, Shields JA, Kwa DM, Arbizo VV. Congenital hypertrophy of the retinal pigment epithelium. Electron microscopic and morphometric observations. Ophthalmology. 1990 Aug:97(8):1052-60 [PubMed PMID: 2402417]
Buettner H. Congenital hypertrophy of the retinal pigment epithelium. American journal of ophthalmology. 1975 Feb:79(2):177-89 [PubMed PMID: 1115190]
Parsons MA, Rennie IG, Rundle PA, Dhingra S, Mudhar H, Singh AD. Congenital hypertrophy of retinal pigment epithelium: a clinico-pathological case report. The British journal of ophthalmology. 2005 Jul:89(7):920-1 [PubMed PMID: 15965180]
Level 3 (low-level) evidenceShields JA, Tso MO. Congenital grouped pigmentation of the retina. Histopathologic description and report of a case. Archives of ophthalmology (Chicago, Ill. : 1960). 1975 Nov:93(11):1153 [PubMed PMID: 1191104]
Level 3 (low-level) evidenceTraboulsi EI, Murphy SF, de la Cruz ZC, Maumenee IH, Green WR. A clinicopathologic study of the eyes in familial adenomatous polyposis with extracolonic manifestations (Gardner's syndrome). American journal of ophthalmology. 1990 Nov 15:110(5):550-61 [PubMed PMID: 2173407]
Gun RD, Akcay G, Kanar HS, Şimşek Ş. From an asymptomatic lesion to a vision-threatening condition: Congenital hypertrophy of the retinal pigment epithelium complicated by choroidal neovascular membrane. Indian journal of ophthalmology. 2020 Oct:68(10):2288-2290. doi: 10.4103/ijo.IJO_2185_19. Epub [PubMed PMID: 32971696]
Augsburger JJ, Henson GL, Hershberger VS, Trichopoulos N. Topographical distribution of typical unifocal congenital hypertrophy of retinal pigment epithelium. Graefe's archive for clinical and experimental ophthalmology = Albrecht von Graefes Archiv fur klinische und experimentelle Ophthalmologie. 2006 Nov:244(11):1412-4 [PubMed PMID: 16568286]
Garoon RB, Harbour JW. Congenital Hypertrophy of the Retinal Pigment Epithelium Presenting With Secondary Choroidal Neovascularization. Ophthalmic surgery, lasers & imaging retina. 2018 Apr 1:49(4):276-277. doi: 10.3928/23258160-20180329-12. Epub [PubMed PMID: 29664987]
Shields JA, Eagle RC Jr, Shields CL, Brown GC, Lally SE. Malignant transformation of congenital hypertrophy of the retinal pigment epithelium. Ophthalmology. 2009 Nov:116(11):2213-6. doi: 10.1016/j.ophtha.2009.04.048. Epub 2009 Sep 10 [PubMed PMID: 19744732]
Level 3 (low-level) evidenceMeyer CH, Rodrigues EB, Mennel S, Schmidt JC, Kroll P. Grouped congenital hypertrophy of the retinal pigment epithelium follows developmental patterns of pigmentary mosaicism. Ophthalmology. 2005 May:112(5):841-7 [PubMed PMID: 15878064]
Meyer CH, Freyschmidt-Paul P, Happle R, Kroll P. Unilateral linear hyperpigmentation of the skin with ipsilateral sectorial hyperpigmentation of the retina. American journal of medical genetics. Part A. 2004 Apr 1:126A(1):89-92 [PubMed PMID: 15039978]
Aiello LP, Traboulsi EI. Pigmented fundus lesions in a preterm infant with familial adenomatous polyposis. Archives of ophthalmology (Chicago, Ill. : 1960). 1993 Mar:111(3):302-3 [PubMed PMID: 8383485]
Traboulsi EI, Krush AJ, Gardner EJ, Booker SV, Offerhaus GJ, Yardley JH, Hamilton SR, Luk GD, Giardiello FM, Welsh SB. Prevalence and importance of pigmented ocular fundus lesions in Gardner's syndrome. The New England journal of medicine. 1987 Mar 12:316(11):661-7 [PubMed PMID: 3821797]
Berk T, Cohen Z, McLeod RS, Parker JA. Congenital hypertrophy of the retinal pigment epithelium as a marker for familial adenomatous polyposis. Diseases of the colon and rectum. 1988 Apr:31(4):253-7 [PubMed PMID: 2896112]
Rodrigues MW, Cavallini DB, Dalloul C, Shields CL, Jorge R. Retinal sensitivity and photoreceptor arrangement changes secondary to congenital simple hamartoma of retinal pigment epithelium. International journal of retina and vitreous. 2019:5():5. doi: 10.1186/s40942-018-0154-7. Epub 2019 Jan 15 [PubMed PMID: 30675383]
Orduña-Azcona J, Gili P, De Manuel-Triantafilo S, Flores-Rodriguez P. Solitary congenital hypertrophy of the retinal pigment epithelium features by high-definition optical coherence tomography. European journal of ophthalmology. 2014 Jul-Aug:24(4):566-9. doi: 10.5301/ejo.5000420. Epub 2013 Dec 20 [PubMed PMID: 24366775]
Level 2 (mid-level) evidenceFrancis JH, Sobol EK, Greenberg M, Folberg R, Abramson DH. Optical Coherence Tomography Characteristics of the Choroid Underlying Congenital Hypertrophy of the Retinal Pigment Epithelium. Ocular oncology and pathology. 2020 Aug:6(4):238-243. doi: 10.1159/000504712. Epub 2020 Feb 27 [PubMed PMID: 33005612]
Touriño R, Rodríguez-Ares MT, López-Valladares MJ, Gómez-Ulla F, Gómez-Torreiro M, Capeans C. Fluorescein angiographic features of the congenital hypertrophy of the retinal pigment epithelium in the familial adenomatous polyposis. International ophthalmology. 2005 Feb-Apr:26(1-2):59-65 [PubMed PMID: 16779566]
Kim DY, Hwang JC, Moore AT, Bird AC, Tsang SH. Fundus autofluorescence and optical coherence tomography of congenital grouped albinotic spots. Retina (Philadelphia, Pa.). 2010 Sep:30(8):1217-22. doi: 10.1097/IAE.0b013e3181cea5a5. Epub [PubMed PMID: 20539258]
Almeida A, Kaliki S, Shields CL. Autofluorescence of intraocular tumours. Current opinion in ophthalmology. 2013 May:24(3):222-32. doi: 10.1097/ICU.0b013e32835f8ba1. Epub [PubMed PMID: 23429597]
Level 3 (low-level) evidenceShanmugam PM, Konana VK, Ramanjulu R, Mishra KCD, Sagar P, Simakurthy S. Ocular coherence tomography angiography features of congenital hypertrophy of retinal pigment epithelium. Indian journal of ophthalmology. 2019 Apr:67(4):563-566. doi: 10.4103/ijo.IJO_801_18. Epub [PubMed PMID: 30900602]
Zucchiatti I, Battaglia Parodi M, Pala M, Bandello FM. Macular congenital hypertrophy of the retinal pigment epithelium: a case report. European journal of ophthalmology. 2010 May-Jun:20(3):621-4 [PubMed PMID: 20155702]
Level 3 (low-level) evidenceMitchem JB, Hall JF. Adenomatous Polyposis Syndromes: Diagnosis and Management. Clinics in colon and rectal surgery. 2016 Dec:29(4):321-329. doi: 10.1055/s-0036-1584089. Epub [PubMed PMID: 31777463]
Tripathy K, Bandyopadhyay G, Basu K, Vatwani KK, Shekhar H. Congenital simple hamartoma of the retinal pigment epithelium with depigmentation at the margin in an Indian female. GMS ophthalmology cases. 2019:9():Doc23. doi: 10.3205/oc000112. Epub 2019 Jun 18 [PubMed PMID: 31355121]
Level 3 (low-level) evidenceChawla R, Kumar V, Tripathy K, Kumar A, Venkatesh P, Shaikh F, Vohra R, Molla K, Verma S. Combined Hamartoma of the Retina and Retinal Pigment Epithelium: An Optical Coherence Tomography-Based Reappraisal. American journal of ophthalmology. 2017 Sep:181():88-96. doi: 10.1016/j.ajo.2017.06.020. Epub 2017 Jun 29 [PubMed PMID: 28669779]
Kumar V,Chawla R,Tripathy K, Omega Sign: A Distinct Optical Coherence Tomography Finding in Macular Combined Hamartoma of Retina and Retinal Pigment Epithelium. Ophthalmic surgery, lasers [PubMed PMID: 28195614]
Geetha R, Tripathy K. Chorioretinitis. StatPearls. 2024 Jan:(): [PubMed PMID: 31869169]
Tripathy K, Sarma B, Mazumdar S. Commentary: Inner retinal excavation in torpedo maculopathy and proposed type 3 lesions in optical coherence tomography. Indian journal of ophthalmology. 2018 Aug:66(8):1213-1214. doi: 10.4103/ijo.IJO_656_18. Epub [PubMed PMID: 30038187]
Level 3 (low-level) evidenceTrichopoulos N, Augsburger JJ, Schneider S. Adenocarcinoma arising from congenital hypertrophy of the retinal pigment epithelium. Graefe's archive for clinical and experimental ophthalmology = Albrecht von Graefes Archiv fur klinische und experimentelle Ophthalmologie. 2006 Jan:244(1):125-8 [PubMed PMID: 15983818]
Mehta N, Gal-Or O, Barbazetto I, Modi Y, Shields CL, Freund KB. ATYPICAL CONGENITAL HYPERTROPHY OF THE RETINAL PIGMENT EPITHELIUM COMPLICATED BY PRESUMED RETINAL PIGMENT EPITHELIAL ADENOMA AND EXUDATIVE MACULOPATHY. Retinal cases & brief reports. 2021 May 1:15(3):330-334. doi: 10.1097/ICB.0000000000000800. Epub [PubMed PMID: 30063581]
Level 3 (low-level) evidenceMoulin AP, Zografos L, Schalenbourg A. RPE adenocarcinoma arising from a congenital hypertrophy of the RPE (CHRPE) treated with proton therapy. Klinische Monatsblatter fur Augenheilkunde. 2014 Apr:231(4):411-3. doi: 10.1055/s-0034-1368287. Epub 2014 Apr 25 [PubMed PMID: 24771179]