Introduction
Beta thalassemia is a group of inherited hemoglobinopathies that can lead to anemia of varying severity.[1] Beta thalassemias are caused by a number of mutations that affect the different aspects of beta globin production, like the transcription, translation, or stability of the beta-globin product. This eventually leads to defective hemoglobin, which is susceptible to destruction.[2]
The diagnosis starts at the clinic, where apparent signs and symptoms can be gathered based on history and examination. The age and ethnic background of the patient are valuable clinical information. To confirm the diagnosis, laboratory testing is a necessary requisite. Laboratory evaluation for beta thalassemia can vary from routine blood tests like peripheral smears, complete blood count, iron studies, and hemoglobin analysis to more complex tests, including genetic testing.[3]
Etiology and Epidemiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology and Epidemiology
Hemoglobin is made of globin proteins, consisting of two alpha and two beta chains bound together by a haem ring. The number of alpha globins must precisely match the number of beta globins. If there is any form of mismatch, it impairs the ability to form normal hemoglobin.[4]
Beta thalassemia is an autosomal recessive disorder caused by a mutation of the Hb B gene located on chromosome number 11, which is responsible for beta-globin synthesis.[1] The clinical presentation depends on genetic abnormality's effect at the molecular level and the combination of other genetic and environmental factors. Beta thalassemia is subdivided into multiple types. This can be beta thalassemia, with no HBB protein produced from the mutated gene(B0), or beta thalassemia, resulting in decreased quantities of protein produced(B+).[2]
Heterozygotes for either type of the allele present with microcytosis clinically named beta thalassemia minor. Compound heterozygotes for two (B+) thalassemia alleles or one B+ and one B0 allele have comparatively more severe symptoms like hemolysis, anemia, iron loading, and at the time, require transfusion. They are clinically referred to as Beta Thalassemia Intermedia. And individuals with both B0 thalassemia alleles present with the most severe form of the disease with transfusion-dependent anemia termed Beta thalassemia major.[2][5]
Beta thalassemia varies widely, generally depending on the ethnic population. It is most common in Africa, the Mediterranean, and Southeast Asian people.[5] The highest carrier frequency is reported in Cyprus, followed by Sardinia and Southeast Asia. It is relatively rare in the United States. The incidence is estimated to be 1 in the 10,000 general population. The migration of people and inter-ethnicity marriages have introduced thalassemia in every part of the world, even in northern Europe, where it was previously absent.[6]
Pathophysiology
The genetic mutation results in a deficiency of the beta-globin chain. The excess unpaired alpha globin leads to hemolysis of Red blood cells in the bone marrow and extramedullary hematopoiesis, causing chronic anemia.[5] As the body tries to compensate, it leads to extramedullary hematopoiesis, which can manifest as skeletal abnormalities in the face and the long bones, hepatosplenomegaly, growth impairment, and kidney enlargement.[7]
The constellation of symptoms, complications, and the degree of anemia may make the patient transfusion-dependent, which can have various complications. Beta thalassemia patients have high iron levels in their Red blood cell membrane due to denatured hemoglobin.[5]
Ineffective erythropoiesis, chronic anemia, and hypoxia can cause increased GI absorption of iron.[8] This, in combination with iron from the destruction of RBCs and repeated transfusions, causes iron deposition in tissues leading to hemosiderosis.[9]
Free iron species like the labile plasma iron and the labile iron pool in RBCs can contribute if transferrin saturation exceeds 70%. These free iron species eventually generate reactive oxygen species, which cause tissue damage and organ dysfunction.[7]
Specimen Requirements and Procedure
The specimen collection varies with the sample chosen for testing. The diagnosis requires a combination of laboratory tests, including measuring red cell indices, Hb analysis, and quantifying HbF and HbA2. Whole blood is collected in EDTA vials, and samples are sent for a complete blood profile, electrophoresis, red cell indices, and molecular testing.[3] EDTA is the preferred anticoagulant, although other anticoagulants can also be used.[10]
Diagnostic Tests
Complete Blood Count
This test can be carried out on an automated hematology analyzer. Red cell counts are usually raised. Microcytic hypochromic anemia is found in thalassemias, except for asymptomatic carriers.[1] Hb, MCV, and MCH cannot differentiate between thalassemia traits, iron deficiency anemia, or alpha and beta thalassemia. Thalassemia does not directly affect the platelet count and white blood count.[3]
If leucocytosis occurs, it may signify an acute infection. However, for unknown reasons, many individuals with thalassemia can have elevated neutrophil counts. Iron deficiency anemia must be ruled out before diagnosing thalassemia.[3][11]
Iron Studies
Normal or slightly increased ferritin levels are present in thalassemia. Transferrin levels are almost normal in thalassemia as compared to iron deficiency anemia.[3]
Peripheral Smear
Peripheral smear shows microcytic hypochromic anemia with target cells, teardrops, and cells with basophilic stipplings.[3] These are non-specific findings that are found in various other anemias as well.
Haemoglobin Analysis
Automated high-performance liquid chromatography and capillary electrophoresis systems are precise and sensitive methods for quantitative and qualitative analyses of Haemoglobin in RBCs.[12]
Molecular Testing
About 200 mutations causing beta thalassemia have been reported, and the most common among them is point mutation.[5] Point mutations are single-base substitutions, minor insertions, or deletions. Some commonly used DNA analysis techniques are Gap-PCR, reverse dot blot (RDB) analysis, real-time PCR with melting curve analysis, and DNA sequencing.[2]
Testing Procedures
For qualitative and quantitative hemoglobin analysis, there are various procedures available. The most widely used among them are electrophoresis and high-performance liquid chromatography.
High-performance Liquid Chromatography
HPLC is used increasingly as the diagnostic procedure for hemoglobinopathies. Its working principle lies behind the interaction between the charges on the ion exchange material and that of the hemoglobin molecule. It has a column packed with silica gel, whose surface is modified by carboxyl charges to give it a weak cationic charge, which permits hemoglobin separation with different charges following the ion exchange phenomena.[13]
The resulting chromatograms are separated by retention time (RT). Retention time and percentages of variant hemoglobin give important clues for differentiating various hemoglobins eluting in the same window. Most importantly, it can quantify the number of various hemoglobins, which help in diagnosis. This method can give false negative results in newborns.[14]
Cellulose Acetate Electrophoresis
This is a simple, reliable, and rapid process where the hemoglobin undergoes electrophoresis at pH 8.4-8.6 using a cellulose acetate membrane. Its principle is that hemoglobin is a negatively charged protein at alkaline pH, and on electrophoresis, it migrates to the anode. The structural variants having a charge on the surface will separate from HbA on electrophoresis.[13] However, hemoglobins with amino acid substitution placed internally or having amino acid causing no change in overall charge may not separate.[14] The accuracy of diagnosis depends on the laboratory personnel's experience and training, and values of HbA2 are not always reproducible.
Microcolumn Chromatography
Microcolumn chromatography depends on the exchange of charged groups between the charged group of the hemoglobin molecule and charged groups on ion exchange cellulose. If a mixture of hemoglobins is adsorbed on the cellulose, particular hemoglobin would be separated from the column using a developer(buffer) at a specific pH or ionic strength. To elute other components, one must change the ionic strength or pH of the developer. The separation of the hemoglobin components is determined by the pH and ionic strength of the developer used, the type of cellulose, the column size, the sample volume, the gradient, temperature, and flow rates.
Molecular Genetic Testing
There is a prevalence of particular mutations in each population. Therefore the origin of individuals plays an important role. The methods for genetic testing are variable and complex. For screening, less expensive multiplex methods are options, like gap PCR, reverse dot blot hybridization, and amplification refractory mutation systems [ARMS].[14]
More complex and exhaustive methods are used for more specific testing, like multiplex ligation probe amplification to detect deletions and Sanger sequencing to look for all possible mutations in an individual. These two methods are capable of revealing unknown mutations.[11]
Supplemental Tests
Some additional tests are necessary for thalassemia patients, which can be classified as iron-related and non-iron-related. Iron metabolism monitoring is essential in these patients as they may have iron overload either due to ineffective erythropoiesis or repeated blood transfusions. Increased iron may get deposited in organs causing endocrine dysfunction and growth retardation.[7]
Therefore routine iron studies are necessary. In addition, Thalassemia patients are susceptible to thrombotic events and hypercoagulability. Therefore tests like PT, aPTT, d-dimer, protein C, coagulation factors, platelet count, and D-dimer should be done. Due to repetitive blood transfusions, they are at increased risk of transfusion-transmitted infective hepatitis, which should be screened in supplement to liver function tests.[15]
Interfering Factors
There are various interfering factors in testing for beta thalassemia. It can occur at any of the steps of diagnosis, including during sample collection, transport, reporting, or internal validation of laboratory results using the standard protocol. The collection, storage, and transport of the sample in anticoagulant and the ratio between the anticoagulant and the blood should be maintained, as a disproportion can cause a discrepancy in results.
In laboratories, the buffers, agents, and stains, if stored improperly, can yield false results and proper slide fixation and staining. Hemoglobin electrophoresis and HPLC can detect other hemoglobins like HbS, Hb C, Hb E, and Hb O, which can interact with beta thalassemia.[14]
In addition, they cannot identify specific variants of beta thalassemia and may miss the diagnosis in newborns. The allele-specific methodologies in gene detection, including PCR and reverse dot-blot, are of limited use in a diverse population.[14]
Results, Reporting, and Critical Findings
In normal adults, Hemoglobin A (HbA) makes up 95 to 98% of the total hemoglobin, hemoglobin A2 (HbA2) is 2 to 3% of hemoglobin, and hemoglobin F(HbF) makes up less than 2% of total adult hemoglobin.[3] An increased Hb A2 is present in heterozygous beta thalassemia; therefore, its precise measurement is essential for diagnosing or excluding the beta thalassemia trait.
Beta Thalssemia Minor
Thalassemia minor is also called beta thalassemia carrier, beta thalassemia trait, and heterozygous beta thalassemia. The most important laboratory tests for diagnosing asymptomatic carriers are red cell count (RBC) and red cell indices.B0 or B++ carriers have increased red blood cell count with decreased MCV (60–70 fl), MCH (19–23 pg) and hemoglobin can range from 2 g/dL to normal.[3]
Reticulocytes may be normal or slightly increased but do not have any significant value in diagnosis. RBCs may show morphological changes like microcytosis, hypochromia, and anisopoikilocytosis. The most valuable test for definite diagnosis is HBA2 quantification by various hemoglobin analysis methods, as described above. The normal value of HBA2 is less than 3.2%, while 3.2 to 3.6% is considered borderline, which warrants further investigation. Values between 3.6% to 7% are considered beta thalassemia carriers. Values of HBA2 are variable in thalassemia. Sometimes iron deficiency anemia may be confused with thalassemia carriers due to microcytosis in both.[5] Therefore, transferrin saturation, Mentzer's index, Shine and Lal Index, or Srivastava index can be used to distinguish the two.[3]
Beta Thalssemia Intermedia
They have mild-moderate anemia with hemoglobin, even in severe forms not going below 7 g/dL, ranging from 7 to 10 g/dL. In thalassemia intermedia, there is more than or equal to 3.5% HbA2, and HbF ranges between 10 to 50%. The molecular defect and the degree of erythropoiesis determine the amount of Hb A2 and Hb F. Molecular testing is a more accurate diagnostic tool.[16]
Beta Thalassemia Major
They present early in life with severe normocytic/microcytic anemia. The hemoglobin level is usually less than 7 g/dL. The MCH is low, <20 pg, and peripheral smear show marked poikilocytosis with tear-drop cell, erythroblasts, and target cells.[3] In classical beta thalassemia major (B0 homozygotes), the hemoglobin analysis shows to be absent, and HbF constitutes 92 to 95% of the total hemoglobin. In major forms resulting from double heterozygosity of B0/B+, the HbA levels are between 10 and 30%, and HbF comprises 70 and 90% of total hemoglobin.[3]
Clinical Significance
Beta thalassemia has a spectrum of clinical presentations. Therefore a precise diagnosis is of paramount importance for optimum patient care. A prenatal diagnosis of beta thalassemia can help prevent the disease. There are non-invasive methods like extraction of DNA from fetal cells in maternal circulation or directly using free fetal DNA in maternal circulation.[17] However, these non-invasive methods require knowing the exact parental haplotype; therefore, they are still not used widely.[18]
Among the invasive methods is obtaining fetal DNA by chorionic villi sampling by ultrasound-guided transabdominal or ultrasound-guided transcervical biopsy. Another invasive method is using amniotic fluid cells to culture fetal DNA. The DNA acquired by amniotic fluid cells is not much, but enough for PCR-based analysis.[19]
Quality Control and Lab Safety
The most important factors for quality are its definition, creation, and control. The errors can be permanent or variable, as well as internal or external.[20] In-house standard operating procedures must be carried out carefully, as a slight change can make a significant difference, leading to erroneous results. Therefore appropriate quality control, along with external quality assurance, is of paramount importance.[21]
It is necessary to use reference standards and control materials in each analysis undertaken and, in some cases, to use duplicate analysis to demonstrate precision. Control materials can be prepared in-house or obtained commercially. All laboratories should confirm the normal range for their particular methods, and the normal range obtained should not differ significantly from the published data.
To check on laboratory tests for any errors or deviation from the normal requires monitoring; one such method of monitoring is the use of the Levey Jennings chart. This uses controls or averages of the results presented graphically. A single-day error or trend can be identified over time, and corrective action can be taken. Additionally, laboratories should also participate in appropriate proficiency testing programs to achieve lab quality assurance.
Enhancing Healthcare Team Outcomes
It is of paramount importance that the clinician understands a detailed workup of a suspected beta thalassemia patient. Every step in diagnosis requires communication, coordination, and responsibility by the staff involved, whether it is storage, processing, or reporting. Regular updates about diagnostic criteria and laboratory techniques must be performed. All the staff members must be aware of the safety precautions required in the lab.
Media
(Click Image to Enlarge)
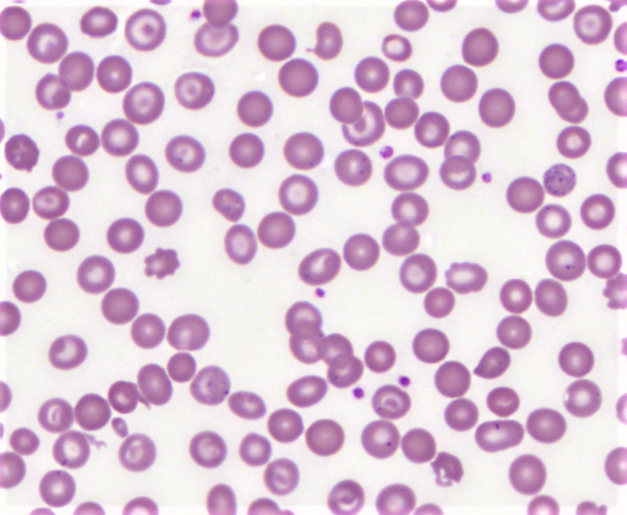
Target cell in Peripheral smear Contributed by Anam Khan
(Click Image to Enlarge)
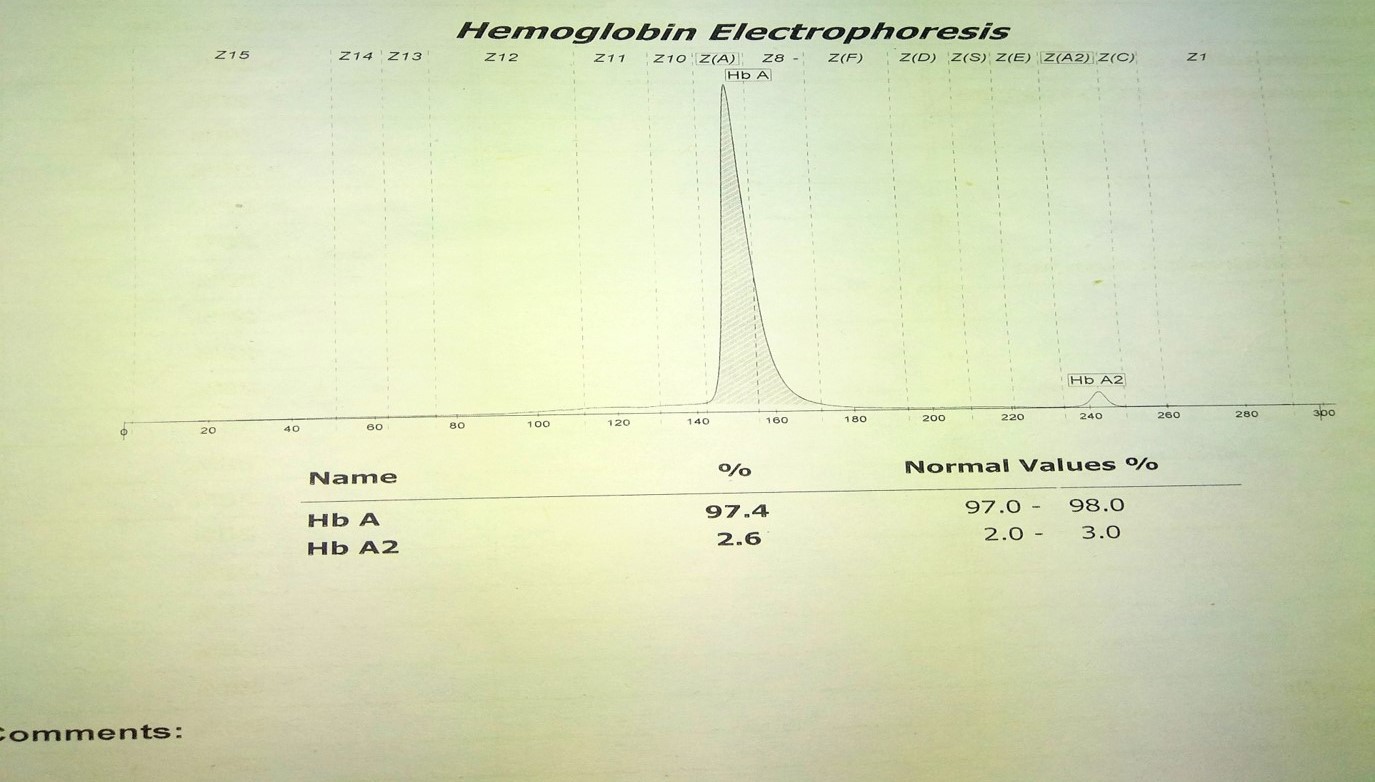
Normal Hemoglobin electrophoresis Contributed by Anam Khan MD
(Click Image to Enlarge)
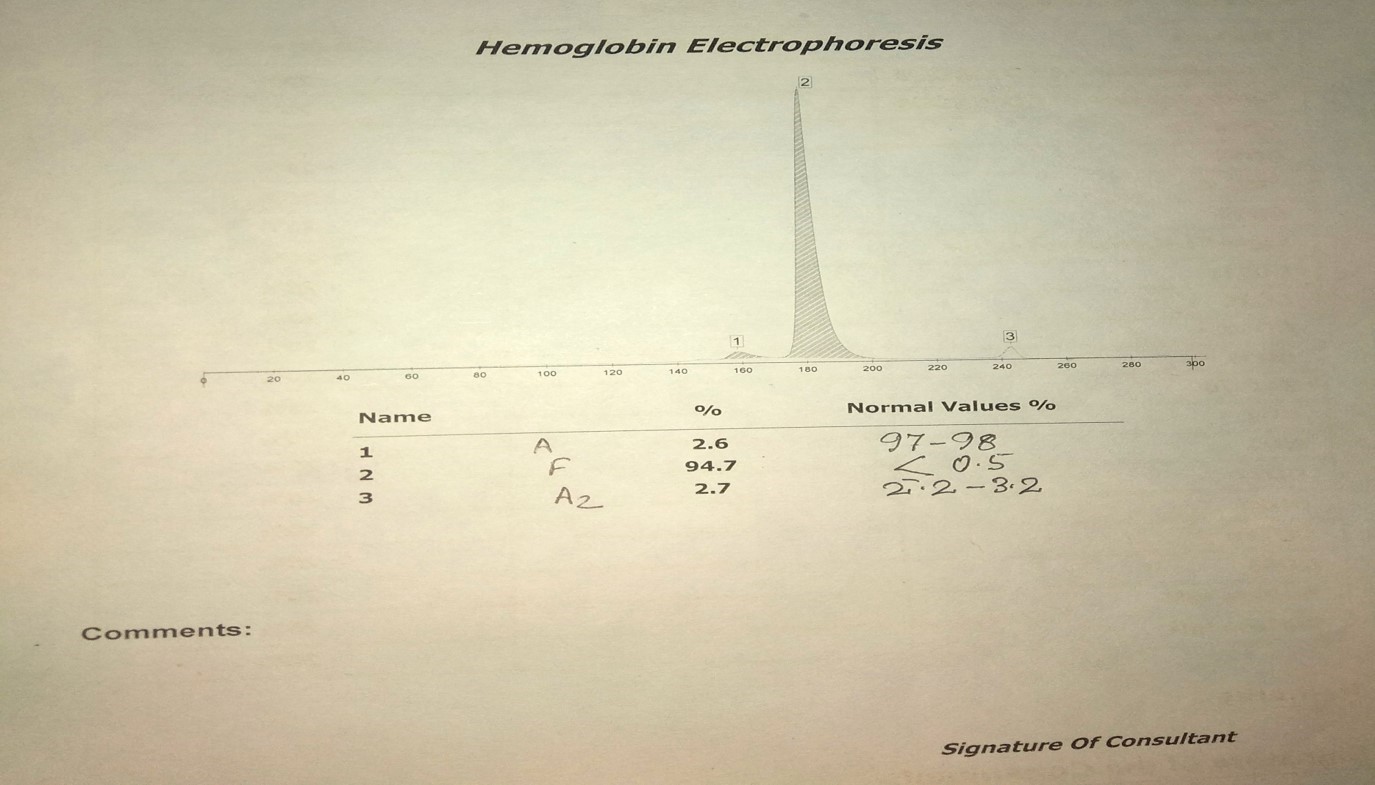
Hemoglobin electrophoresis showing Beta thalassemia Major Contributed by Anam Khan MD
References
Galanello R, Origa R. Beta-thalassemia. Orphanet journal of rare diseases. 2010 May 21:5():11. doi: 10.1186/1750-1172-5-11. Epub 2010 May 21 [PubMed PMID: 20492708]
Lee JS, Cho SI, Park SS, Seong MW. Molecular basis and diagnosis of thalassemia. Blood research. 2021 Apr 30:56(S1):S39-S43. doi: 10.5045/br.2021.2020332. Epub [PubMed PMID: 33935034]
Brancaleoni V, Di Pierro E, Motta I, Cappellini MD. Laboratory diagnosis of thalassemia. International journal of laboratory hematology. 2016 May:38 Suppl 1():32-40. doi: 10.1111/ijlh.12527. Epub 2016 May 16 [PubMed PMID: 27183541]
Farid Y, Bowman NS, Lecat P. Biochemistry, Hemoglobin Synthesis. StatPearls. 2023 Jan:(): [PubMed PMID: 30725597]
Olivieri NF. The beta-thalassemias. The New England journal of medicine. 1999 Jul 8:341(2):99-109 [PubMed PMID: 10395635]
Weatherall DJ. The inherited diseases of hemoglobin are an emerging global health burden. Blood. 2010 Jun 3:115(22):4331-6. doi: 10.1182/blood-2010-01-251348. Epub 2010 Mar 16 [PubMed PMID: 20233970]
Level 2 (mid-level) evidenceRivella S. Ineffective erythropoiesis and thalassemias. Current opinion in hematology. 2009 May:16(3):187-94. doi: 10.1097/MOH.0b013e32832990a4. Epub [PubMed PMID: 19318943]
Level 3 (low-level) evidenceLongo F, Piolatto A, Ferrero GB, Piga A. Ineffective Erythropoiesis in β-Thalassaemia: Key Steps and Therapeutic Options by Drugs. International journal of molecular sciences. 2021 Jul 5:22(13):. doi: 10.3390/ijms22137229. Epub 2021 Jul 5 [PubMed PMID: 34281283]
Hershko C, Rachmilewitz EA. Mechanism of desferrioxamine-induced iron excretion in thalassaemia. British journal of haematology. 1979 May:42(1):125-32 [PubMed PMID: 465354]
Fakher R, Bijan K, Taghi AM. Application of diagnostic methods and molecular diagnosis of hemoglobin disorders in Khuzestan province of Iran. Indian journal of human genetics. 2007 Jan:13(1):5-15. doi: 10.4103/0971-6866.32028. Epub [PubMed PMID: 21957335]
Traeger-Synodinos J, Harteveld CL. Advances in technologies for screening and diagnosis of hemoglobinopathies. Biomarkers in medicine. 2014:8(1):119-31. doi: 10.2217/bmm.13.103. Epub [PubMed PMID: 24325233]
Level 3 (low-level) evidenceOyaert M, Van Laer C, Claerhout H, Vermeersch P, Desmet K, Pauwels S, Kieffer D. Evaluation of the Sebia Minicap Flex Piercing capillary electrophoresis for hemoglobinopathy testing. International journal of laboratory hematology. 2015 Jun:37(3):420-5. doi: 10.1111/ijlh.12305. Epub 2014 Oct 16 [PubMed PMID: 25324031]
Colah RB, Surve R, Sawant P, D'Souza E, Italia K, Phanasgaonkar S, Nadkarni AH, Gorakshakar AC. HPLC studies in hemoglobinopathies. Indian journal of pediatrics. 2007 Jul:74(7):657-62 [PubMed PMID: 17699975]
Sabath DE. Molecular Diagnosis of Thalassemias and Hemoglobinopathies: An ACLPS Critical Review. American journal of clinical pathology. 2017 Jul 1:148(1):6-15. doi: 10.1093/ajcp/aqx047. Epub [PubMed PMID: 28605432]
Singh S, Yadav G, Kushwaha R, Jain M, Ali W, Verma N, Verma SP, Singh US. Bleeding Versus Thrombotic Tendency in Young Children With Beta-Thalassemia Major. Cureus. 2021 Dec:13(12):e20192. doi: 10.7759/cureus.20192. Epub 2021 Dec 6 [PubMed PMID: 34877233]
Origa R, Baldan A, Marsella M, Borgna-Pignatti C. A complicated disease: what can be done to manage thalassemia major more effectively? Expert review of hematology. 2015 Dec:8(6):851-62. doi: 10.1586/17474086.2015.1101339. Epub 2015 Oct 15 [PubMed PMID: 26470003]
Madgett TE. First Trimester Noninvasive Prenatal Diagnosis of Maternally Inherited Beta-Thalassemia Mutations. Clinical chemistry. 2022 Jul 27:68(8):1002-1004. doi: 10.1093/clinchem/hvac103. Epub [PubMed PMID: 35757992]
Chen C, Li R, Sun J, Zhu Y, Jiang L, Li J, Fu F, Wan J, Guo F, An X, Wang Y, Fan L, Sun Y, Guo X, Zhao S, Wang W, Zeng F, Yang Y, Ni P, Ding Y, Xiang B, Peng Z, Liao C. Noninvasive prenatal testing of α-thalassemia and β-thalassemia through population-based parental haplotyping. Genome medicine. 2021 Feb 5:13(1):18. doi: 10.1186/s13073-021-00836-8. Epub 2021 Feb 5 [PubMed PMID: 33546747]
Agarwal S, Gupta A, Gupta UR, Sarwai S, Phadke S, Agarwal SS. Prenatal diagnosis in beta-thalassemia: an Indian experience. Fetal diagnosis and therapy. 2003 Sep-Oct:18(5):328-32 [PubMed PMID: 12913343]
Petersen PH, Ricós C, Stöckl D, Libeer JC, Baadenhuijsen H, Fraser C, Thienpont L. Proposed guidelines for the internal quality control of analytical results in the medical laboratory. European journal of clinical chemistry and clinical biochemistry : journal of the Forum of European Clinical Chemistry Societies. 1996 Dec:34(12):983-99 [PubMed PMID: 8986407]
Level 2 (mid-level) evidenceBadrick T. Integrating quality control and external quality assurance. Clinical biochemistry. 2021 Sep:95():15-27. doi: 10.1016/j.clinbiochem.2021.05.003. Epub 2021 May 7 [PubMed PMID: 33965412]
Level 2 (mid-level) evidence