Introduction
The term Alport syndrome is used to describe a group of heterogeneous inherited disorders involving the basement membrane of the kidney and commonly affecting the cochlea and eye.[1][2] Alport syndrome, also known as hereditary nephritis, is a genetic disorder arising from the mutations in the genes encoding alpha-3, alpha-4, and alpha-5 of type 4 collagen (COL4A3, COL4A4, COL4A5) or collagen 4 α345 network.[3]
The type 4 collagen alpha chains are primarily located in the kidneys, eyes, and cochlea. Alport syndrome is X-linked (XLAS) and can be transmitted in an autosomal recessive (ARAS) or autosomal dominant fashion (ADAS). In 1927, the syndrome of hereditary nephritis and deafness was described by a British physician, A. Cecil Alport. It was observed that hematuria was the most common symptom, and males were affected more than females. In 1961, it was named Alport syndrome after having been observed in multiple family members.[4][5][6] It is characterized by renal failure, bilateral sensorineural hearing loss, and eye abnormalities. Eventually, the patients present with proteinuria, hypertension, progressive loss of kidney function (gradual decline in GFR), and end-stage renal disease (ESRD).[7]
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
In 80% of cases, Alport syndrome is inherited in an X-linked pattern and caused by COL4A5 gene mutations, although other inheritance patterns exist. It can be inherited as an autosomal recessive or dominant pattern by mutations in COL4A3 or COL4A4 gene. Approximately 80% of men with XLAS develop some hearing loss until they reach their teenage years.[8]
Alport syndrome is caused by mutations in the genes responsible for alpha-3, alpha-4, and alpha-5 chains of type IV collagen. The following three genetic forms of Alport syndrome are mentioned with their proximate causes:
-
XLAS - caused by mutations in the COL4A5 gene; 85% of cases of Alport syndrome[9]
-
ARAS - due to mutations in COL4A3 or COL4A4 gene; leading to approximately 10-15% of cases
-
ADAS - caused by mutations in the COL4A3 or COL4A4 gene, although rare
Over 300 mutations have been observed in the COL4A5 genes in families with XLAS. Majorly, COL4A5 mutations are minor, including splice-site mutations, missense mutations, and deletions of fewer than ten base pairs.
Around 20% of the mutations are large and medium-sized deletions in the COL4A5 gene. A certain kind of deletion involving the COL4A5 and COL4A6 genes leads to a combination of XLAS and leiomyomatosis of the esophagus, female genital tract, and tracheobronchial tree.[10]
In patients with Alport syndrome, to date, only six mutations in the COL4A3 gene and twelve mutations in the COL4A4 gene are seen in patients with ARAS. The mutations include frameshift deletions, amino acid substitutions, missense mutations, splicing mutations, and in-frame deletions.
Epidemiology
Alport syndrome affects about 1 in 50,000 newborns, and males are more likely to be symptomatic than females. It is estimated that approximately 30,000 to 60,000 people in the United States (US) have this disorder. In the US, the overall incidence of end-stage renal disease (ESRD) in children is about 3% and 0.2% in the adult population.[11][12]
Alport syndrome accounts for approximately 2.2% of children and 0.2% of adults with ESRD in the United States. In Europe, Alport syndrome accounts for 0.6% of patients with ESRD.
The common X-linked type of Alport syndrome resulting in ESRD predominantly affects males. However, the X-linked form of Alport syndrome affects almost as many females. Affected women are generally undiagnosed, but 15%–30% end up having renal failure by 60 years and hearing loss by middle age.[13]
Alport syndrome is a significant cause of chronic kidney disease (CKD), leading to ESRD in adolescents and young adults, accounting for 1.5% to 3.0% of children on renal replacement therapies in Europe and the US.[14]
Pathophysiology
The pathophysiology of Alport syndrome is impaired production and deposition of collagen 4 α345 network in the basement membranes of the glomerulus, cochlea (inner ear), and eye. ARAS transmission is due to mutations in both the alleles of COL4A3 and COL4A4, whereas ADAS is caused by heterozygous mutations. With the use of next-generation sequencing (NGS), it has been shown that ADAS accounts for a greater number of cases. Compared to XLAS, patients with ADAS have a slower rate of progression to ESRD and are less likely to have extra-renal manifestations.[15][16] The glomerular basement membrane (GBM) in Alport syndrome is more prone to proteolytic injury leading to activation of adhesion kinase in podocytes, endothelin receptors, glomerular inflammation, tubulointerstitial fibrosis, and ESRD.[17][18][19][20]
The primary pathology in patients with Alport syndrome—causing abnormality in the basement membrane—is present in the noncollagenous (NC1) C-terminal of the alpha-5 chain in XLAS and that of alpha-3 or alpha-4 chains in ARAS and ADAS.
In the early development of the kidney, alpha-1 and alpha-2 chains are predominant in the glomerular basement membrane (GBM). With maturation, alpha-3, alpha-4, and alpha-5 chains become dominant through isotype switching. Evidence suggests that alpha-3, alpha-4, and alpha-5 chains combine to produce a unique collagen network. Abnormalities in any of these chains, as seen in patients with Alport syndrome, impair the formation of the collagen network leading to aberrant incorporation of the other collagen chains.
Evidence shows that isoform switching of type IV collagen is developmentally arrested in XLAS.[21] This causes the perpetuation of the fetal distribution of alpha-1 and alpha-2 isoforms and a complete lack of alpha-3, alpha-4, and alpha-5 isoforms. The anomalous retention of alpha-1 and alpha-2 isoforms makes GBM prone to proteolytic enzymes, causing basement membrane splitting and damage.
Patients who develop anti-GBM nephritis have circulating anti-GBM antibodies. In patients with ARAS, antibodies bind to the alpha-3 and alpha-4 collagen chains, whereas in patients with XLAS, antibodies predominantly bind to the alpha-5 chain.[22][23][24] Native kidneys do not express the antigens recognized by the anti-GBM antibodies but are expressed in the transplanted kidneys, which are recognized by these antibodies.
Posttransplant anti-GBM nephritis generally appears within the first year of transplant. Patients have rapidly progressive glomerulonephritis with kidney biopsy revealing linear immune deposits along the GBM and crescentic glomerulonephritis. Contrary to de novo anti-GBM nephritis, lung hemorrhage is never seen in posttransplant anti-GBM nephritis in cases of Alport syndrome. Plasmapheresis and cyclophosphamide are generally unsuccessful, and most patients lose the allograft.[25] However, there have been cases of successful treatment with intravenous immunoglobulins and plasmapheresis.
Histopathology
Initially, kidney biopsy specimens examined through light microscopy may be normal. As the disease progresses, non-specific findings may appear. These include focal and segmental glomerulosclerosis, tubular atrophy, interstitial fibrosis, and the presence of lymphocytes and plasma cells. Immunofluorescence studies yield negative results. Electron microscopy of the kidney reveals longitudinal splitting and multi-lamellation of the GBM lamina densa.[26] Monoclonal antibodies against alpha-3, alpha-4, and alpha-5 chains of type IV collagen may be used to assess the glomerular basement membrane for the presence or absence of these collagen chains. Their absence is strongly suggestive of Alport syndrome and is not seen in any other condition.
As the alpha-5 chain of type 4 collagen is also expressed in the epidermis, a skin biopsy can be used to establish a diagnosis. Immunofluorescence studies on skin biopsy specimens are often diagnostic. Patients with XLAS may also display abnormalities of alpha-2 collagen expression in the skin.[27][28]
History and Physical
A thorough history and physical examination must be obtained along with a family history. Laboratory evaluation should include urinalysis (UA), urine microscopy, and renal function panel. Individuals with Alport syndrome may develop symptoms of hematuria, proteinuria, edema, hypertension, progressive decline in renal functions, and eventual ESRD. Over time, the symptoms worsen, and the patients experience worsening proteinuria, hypertension, a decline in GFR, and the development of ESRD. The time for the development of ESRD is around 16 to 35 years of age. They can also present with gross hematuria following an upper respiratory tract infection.[29]
During late childhood, people with Alport syndrome frequently develop bilateral sensorineural hearing loss caused by abnormalities of type 4 collagen in the inner ear.[30] Hearing loss becomes apparent in late childhood or early adolescence, usually before the onset of kidney failure, and starts with high-frequency loss.
Multiple ocular findings can be seen in patients with Alport syndrome. Affected individuals may have a cone-shaped lens (anterior lenticonus), leading to abnormal refraction and decreased visual acuity. Other abnormalities include subcapsular cataracts, abnormal pigmentary changes in the retina with yellow or white flecks (dot-and-fleck retinopathy), posterior polymorphous dystrophy, and corneal erosions.[31][32][33][34]
The following are the features in the history and examination, system-wise:
Renal
-
Gross or microscopic hematuria is the commonest and earliest sign of Alport syndrome.[35] It is more commonly seen in males. Like immunoglobulin, A nephropathy, around 60-70% of patients have episodes of gross hematuria, commonly precipitated by upper respiratory infection, in the first two decades of life. Hematuria is typically discovered in the first years of life in male patients. If there is no hematuria in a male patient in the first decade of life, he is less likely to have Alport syndrome.
-
Proteinuria is generally absent in childhood but ultimately develops in males with XLAS and male and female patients with ARAS.[36]
-
Incidence and severity increase with age and the extent of kidney failure.
-
Hypertension develops by the second decade of life.
-
Edema and nephrotic syndrome are observed in 30 to 40% of young patients with Alport syndrome; however, they are uncommon in early childhood.
-
With the onset of kidney insufficiency, chronic anemia and osteodystrophy may manifest.
Hearing
- Sensorineural hearing loss is a characteristic feature observed commonly, but not universally, in Alport syndrome. Hearing loss is never congenital. Bilateral, high-frequency sensorineural deafness usually begins in late childhood or early adolescence, before the onset of kidney failure. Audiometry is an important tool to diagnose hearing loss at the beginning of the disease.
- As hearing loss progresses, patients require hearing aids.
- Hearing loss is always associated with renal involvement.
- Approximately 50% of male patients with XLAS have sensorineural hearing impairment by the age of 25 years, and around 90% are deaf by 40 years.
Ocular
- Anterior lenticonus, occurring in around 25% of patients with XLAS, is a characteristic feature of Alport syndrome. In this condition, the lens conically protrudes into the anterior chamber due to a thin basement membrane of the lens capsule.
- Anterior lenticonus is generally not present at birth and is manifested by a slow worsening of vision, needing patients to change the prescription of their glasses frequently.
- Anterior lenticonus is diagnosed by slit lamp examination.
- Ultrastructure assessment of the anterior lens capsule with the help of electron microscopy confirms Alport syndrome.[37]
- Dot-and-fleck retinopathy is the commonest ocular manifestation of Alport syndrome, occurring in around 85% of males with XLAS. Rarely seen in childhood, it typically appears at the onset of kidney failure. It is usually asymptomatic. Numerous white and yellow dots and flecks occur around the macula in this condition. There is foveal sparing, but peripheries can get involved. Typically, dot-and-fleck retinopathy will not fluoresce with angiography.
- Posterior polymorphous corneal dystrophy is rare in Alport syndrome. Mostly, patients are asymptomatic, but some may develop gradually progressive visual impairment. A rare feature of Alport syndrome, it manifests as clear vesicles on the endothelial surface of the cornea. It is generally bilateral but can be unilateral.
- The mutation in the COL4A5 gene rarely causes severe temporal macular thinning, a feature associated with XLAS.[38]
- Giant macular holes have been reported in Alport syndrome case reports. The very large dimensions of macular holes were suggestive of a bad prognosis regarding the closure of the hole.[39]
Leiomyomatosis
- Diffuse leiomyomatosis of the tracheobronchial tree and esophagus has been observed in some families with Alport syndrome.[40] It generally becomes symptomatic in late childhood, and symptoms include dysphagia, postprandial vomiting, recurrent bronchitis, substernal or epigastric pain, dyspnea, stridor, and cough. Leiomyomatosis is confirmed on a computed tomography scan or magnetic resonance imaging.
Evaluation
Patients typically present with persistent microscopic hematuria before the age of ten years. This is due to the defective glomerular basement membrane (GBM) permitting the passage of red blood cells.[41] The clinical suspicion for Alport syndrome should be high in patients with hematuria, proteinuria, abnormal renal indices, and ear and eye manifestations. UA would reveal blood and protein, and urine microscopy should be performed to evaluate for acanthocytes.
Renal biopsy is indicated in the setting of abnormal UA, presence of acanthocytes, red blood cell casts, or abnormal renal indices. Any patient with suspected Alport syndrome should be referred to otorhinolaryngology for the evaluation of high-frequency hearing loss and ophthalmology for the eye examination. Due to the defective collagen, the lens lacks the integrity to maintain the normal shape leading to the anterior lenticonus into the anterior chamber.[42][43]
Genetic testing can help establish the diagnosis and determine the inheritance pattern of an individual and their family members. Molecular genetic testing is non-invasive, accurate, and gives the prognosis as the underlying mutation can be revealed. Next-generation sequencing (NGS) analyses of COL4A3, COL4A4, and COL4A5 are recommended in patients with no family history of Alport syndrome.[15][44] For patients with positive family history, testing the target gene is recommended. A renal biopsy is preferred if the genetic defect does not match the family genetic mutation. In males with XLAS, the GBM splitting increases from approximately 30% to more than 90% by the age of 30 years.[45]
Patients without the characteristic GBM splitting can be identified by immunostaining the type 4 collagen of the alpha-3, alpha-4, and alpha-5 chains of the GBM. A less invasive procedure, a skin biopsy can be performed on a child with suspected XLAS using a monoclonal antibody against the alpha-5 of the type 4 collagen chain.
Treatment / Management
Unfortunately, there is no specific treatment for Alport syndrome. Treatment is focused on limiting the progression of proteinuria and kidney disease. Options include angiotensin-converting enzyme inhibitors (ACEi) and angiotensin receptor blockers (ARBs) for managing proteinuria, hypertension, and CKD. Depending upon the degree of proteinuria, diuretics can be used. Although the treatment may delay the onset of renal impairment, most people affected by Alport will ultimately require dialysis or a kidney transplant.[46][47][1]
ARB delays the progression of CKD or ESRD by reducing intra-glomerular pressure and proteinuria. Despite normal renal functions, initiating therapy with ARBs has been shown to have a significant impact on the development of ESRD.[48](B2)
A small study found that combination therapy with an ARB, an ACE inhibitor, a statin, and a non-dihydropyridine calcium channel blocker safely ameliorated hypertension, albuminuria, lipid abnormalities, and glomerular selectivity in patients with Alport syndrome and halted progression in those without renal insufficiency.[49]
A multicenter, placebo-controlled, randomized, double-blind trial of ramipril in children having Alport syndrome observed a decrease in the risk of disease progression by nearly half.[50](A1)
The use of cyclosporine has not shown any benefit and is not recommended. For patients with ocular involvement, specifically anterior lenticonus, clear lens phacoemulsification with intraocular lens implantation can be considered. For patients with concomitant hearing loss, hearing aids are usually very effective. Hearing loss is not impacted by kidney transplantation. As with any hereditary disease, psychosocial support for all affected family members is essential.[51]
Patients with Alport syndrome have no contraindication for renal transplantation. There is no recurrence of the primary disease, as the transplanted kidney would have normal GBM. Antibodies against COL4A5 are found in males with XLAS, but very few patients also have antibodies against COL4A3.[52][53][54][55](B3)
Post renal transplantation, there is a 3% risk of de novo anti-GBM antibody disease or Alport posttransplant nephritis in XLAS with COL4A3. Generally, it relapses in the first year of transplantation.[32][56][57]
The affected patients typically possess circulating anti-GBM antibodies leading to crescentic glomerulonephritis and graft loss.[58][59] Patients with ADAS do not appear to be at increased risk for de novo anti-GBM disease following kidney transplantation. The treatment of post-transplant anti-GBM disease involves plasmapheresis along with cyclophosphamide and methylprednisolone with minimal benefit. Retransplantation in these patients has a high risk of recurrence.(B3)
Differential Diagnosis
The differential diagnoses of Alport syndrome include the following:
-
Immunoglobulin A nephropathy
-
Thin GBM disease
-
Acute post-streptococcal glomerulonephritis
-
Medullary cystic disease
-
Multicystic renal dysplasia
-
Polycystic kidney disease
The most important diagnostic consideration in patients with Alport syndrome is thin basement membrane (TBM) disease, a collagen 4-related nephropathy closely related to Alport syndrome. The same genes appear to be involved in many individuals with the disorder. Unlike those with Alport syndrome, few extra-renal findings are present, symptoms are less severe, with progression to renal impairment is rarely found. Differentiating these disease processes is challenging, particularly in younger or female patients who are less likely to have other associated symptoms.[60]
Prognosis
In the X-linked disease form, the most common type of Alport syndrome, about 50% of males require dialysis or kidney transplantation by 30 years, and approximately 90% develop ESRD before 40.[61] Female patients with X-linked Alport syndrome have a better prognosis, with about 12% developing end-stage renal disease (ESRD) by age 40. However, studies indicate significant renal morbidity in females with proteinuria and hearing impairment.[62][63]
By age 60, this rate increases to about 30%, and by 60 years of age, the rate of ESRD approaches 40%. In the female population, proteinuria and hearing loss are risk factors for the progression to ESRD. In comparison, the autosomal recessive form of Alport syndrome can cause kidney failure by age 20. In contrast, the autosomal dominant form of the disease typically has a delay in ESRD until middle age.[64]
Complications
Alport syndrome affects multiple organ systems. It can lead to the following complications:
- End-stage renal disease (ESRD)
- Hearing loss
- Visual defects
- Leiomyomatosis (smooth muscle overgrowth in the respiratory and gastrointestinal tract)
- Aneurysms of the thoracic and abdominal aorta
- Mental retardation
Consultations
Alport syndrome is managed through a multidisciplinary approach as multiple organ systems are involved. An internist, intensivist, nephrologist, otorhinolaryngologist, ophthalmologist, and geneticist are involved in treating and managing Alport syndrome.
Consultations with the following may also be necessary:
- Transplant surgeon
- Surgeon
- Dialysis specialist
- Audiologist
Deterrence and Patient Education
Patients should be given information leaflets regarding symptoms of renal failure and its systemic effects. Parents of affected children should be knowledgeable about the signs and symptoms of the disease that warrant hospital admission. Family members of patients with Alport syndrome should consult their primary care providers to go through screening as per the local guidelines. Patients with Alport syndrome should undergo genetic counseling before getting married and having children. There are various support groups available that can help patients and their families have better handling of their circumstances.
Enhancing Healthcare Team Outcomes
Alport syndrome is a genetic disorder that affects multiple organs. The condition is best managed by an interprofessional team that includes a geneticist, nephrologist, ophthalmologist, otolaryngologist, and internist/pediatrician. Nurses and pharmacists are also part of this interprofessional team, counseling patients and working with their medications. In most cases, the disorder presents during the first decade of life. Once the diagnosis is made, the workup of siblings and other family members is recommended. Within the first three decades of life, most Alport syndrome patients develop ESRD and need dialysis. Here again, nurses and pharmacists will play a prominent role, the former in administering dialysis and the latter in fine-tuning drug dosing regimens around dialysis. All care team members must exercise open communication with the rest of the team and maintain accurate, updated records to ensure open information sharing among all professionals on the case.
Collaboration and communication among the interprofessional team to enhance care coordination for patients suffering from Alport syndrome is crucial to optimal patient outcomes. [Level 5]
Media
(Click Image to Enlarge)
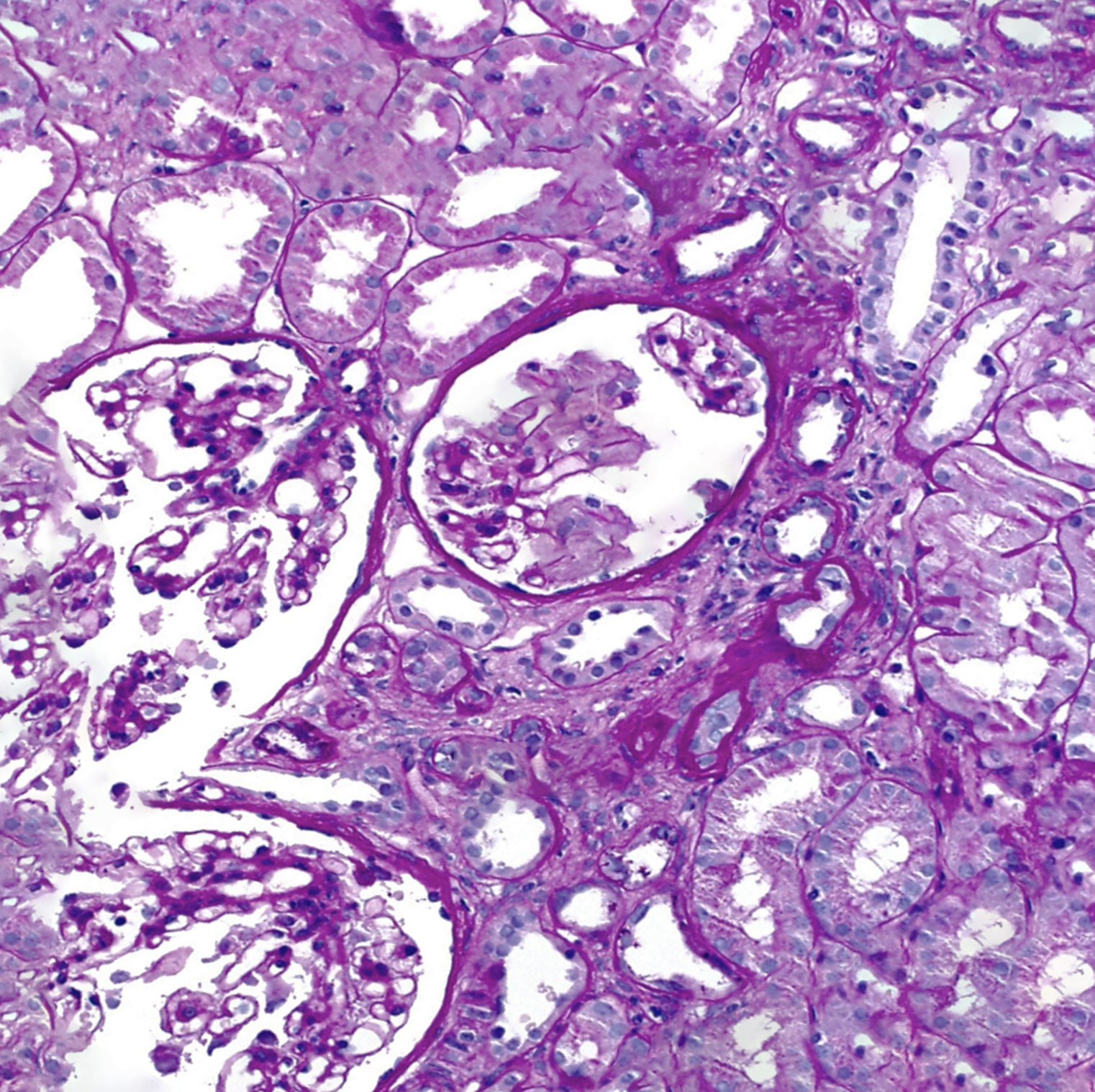
Kidney biopsy showing glomeruli in Alport syndrome. Contributed by Rian Kabir, MD
References
Zhang Y, Ding J. Renal, auricular, and ocular outcomes of Alport syndrome and their current management. Pediatric nephrology (Berlin, Germany). 2018 Aug:33(8):1309-1316. doi: 10.1007/s00467-017-3784-3. Epub 2017 Sep 1 [PubMed PMID: 28864840]
Watson S, Padala SA, Hashmi MF, Bush JS. Alport Syndrome. StatPearls. 2024 Jan:(): [PubMed PMID: 29262041]
Kashtan C. Alport syndrome: facts and opinions. F1000Research. 2017:6():50. doi: 10.12688/f1000research.9636.1. Epub 2017 Jan 17 [PubMed PMID: 28163907]
Level 3 (low-level) evidenceSavige J. Alport syndrome: deducing the mode of inheritance from the presence of haematuria in family members. Pediatric nephrology (Berlin, Germany). 2020 Jan:35(1):59-66. doi: 10.1007/s00467-018-4121-1. Epub 2018 Nov 30 [PubMed PMID: 30506145]
Kashtan CE. Renal transplantation in patients with Alport syndrome: patient selection, outcomes, and donor evaluation. International journal of nephrology and renovascular disease. 2018:11():267-270. doi: 10.2147/IJNRD.S150539. Epub 2018 Oct 16 [PubMed PMID: 30410383]
Vos P, Zietse R, van Geel M, Brooks AS, Cransberg K. Diagnosing Alport Syndrome: Lessons from the Pediatric Ward. Nephron. 2018:140(3):203-210. doi: 10.1159/000492438. Epub 2018 Sep 13 [PubMed PMID: 30212818]
Katsuma A, Nakada Y, Yamamoto I, Horita S, Furusawa M, Unagami K, Katsumata H, Okumi M, Ishida H, Yokoo T, Tanabe K, Japan Academic Consortium of Kidney Transplantation (JACK). Long-term survival in Japanese renal transplant recipients with Alport syndrome: a retrospective study. BMC nephrology. 2018 Oct 3:19(1):249. doi: 10.1186/s12882-018-1052-9. Epub 2018 Oct 3 [PubMed PMID: 30285655]
Level 2 (mid-level) evidenceCrockett DK, Pont-Kingdon G, Gedge F, Sumner K, Seamons R, Lyon E. The Alport syndrome COL4A5 variant database. Human mutation. 2010 Aug:31(8):E1652-7. doi: 10.1002/humu.21312. Epub [PubMed PMID: 20574986]
Hashimura Y, Nozu K, Kaito H, Nakanishi K, Fu XJ, Ohtsubo H, Hashimoto F, Oka M, Ninchoji T, Ishimori S, Morisada N, Matsunoshita N, Kamiyoshi N, Yoshikawa N, Iijima K. Milder clinical aspects of X-linked Alport syndrome in men positive for the collagen IV α5 chain. Kidney international. 2014 May:85(5):1208-13. doi: 10.1038/ki.2013.479. Epub 2013 Dec 4 [PubMed PMID: 24304881]
Level 2 (mid-level) evidenceNozu K, Minamikawa S, Yamada S, Oka M, Yanagita M, Morisada N, Fujinaga S, Nagano C, Gotoh Y, Takahashi E, Morishita T, Yamamura T, Ninchoji T, Kaito H, Morioka I, Nakanishi K, Vorechovsky I, Iijima K. Characterization of contiguous gene deletions in COL4A6 and COL4A5 in Alport syndrome-diffuse leiomyomatosis. Journal of human genetics. 2017 Jul:62(7):733-735. doi: 10.1038/jhg.2017.28. Epub 2017 Mar 9 [PubMed PMID: 28275241]
Hicks J, Mierau G, Wartchow E, Eldin K. Renal diseases associated with hematuria in children and adolescents: a brief tutorial. Ultrastructural pathology. 2012 Feb:36(1):1-18. doi: 10.3109/01913123.2011.620731. Epub [PubMed PMID: 22292732]
Warady BA, Agarwal R, Bangalore S, Chapman A, Levin A, Stenvinkel P, Toto RD, Chertow GM. Alport Syndrome Classification and Management. Kidney medicine. 2020 Sep-Oct:2(5):639-649. doi: 10.1016/j.xkme.2020.05.014. Epub 2020 Aug 7 [PubMed PMID: 33094278]
Savige J, Colville D, Rheault M, Gear S, Lennon R, Lagas S, Finlay M, Flinter F. Alport Syndrome in Women and Girls. Clinical journal of the American Society of Nephrology : CJASN. 2016 Sep 7:11(9):1713-1720. doi: 10.2215/CJN.00580116. Epub 2016 Jun 10 [PubMed PMID: 27287265]
Deltas C, Pierides A, Voskarides K. Molecular genetics of familial hematuric diseases. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association. 2013 Dec:28(12):2946-60. doi: 10.1093/ndt/gft253. Epub 2013 Sep 17 [PubMed PMID: 24046192]
Fallerini C, Dosa L, Tita R, Del Prete D, Feriozzi S, Gai G, Clementi M, La Manna A, Miglietti N, Mancini R, Mandrile G, Ghiggeri GM, Piaggio G, Brancati F, Diano L, Frate E, Pinciaroli AR, Giani M, Castorina P, Bresin E, Giachino D, De Marchi M, Mari F, Bruttini M, Renieri A, Ariani F. Unbiased next generation sequencing analysis confirms the existence of autosomal dominant Alport syndrome in a relevant fraction of cases. Clinical genetics. 2014 Sep:86(3):252-7. doi: 10.1111/cge.12258. Epub 2013 Oct 17 [PubMed PMID: 24033287]
Level 3 (low-level) evidenceMorinière V, Dahan K, Hilbert P, Lison M, Lebbah S, Topa A, Bole-Feysot C, Pruvost S, Nitschke P, Plaisier E, Knebelmann B, Macher MA, Noel LH, Gubler MC, Antignac C, Heidet L. Improving mutation screening in familial hematuric nephropathies through next generation sequencing. Journal of the American Society of Nephrology : JASN. 2014 Dec:25(12):2740-51. doi: 10.1681/ASN.2013080912. Epub 2014 May 22 [PubMed PMID: 24854265]
Level 2 (mid-level) evidenceDelimont D, Dufek BM, Meehan DT, Zallocchi M, Gratton MA, Phillips G, Cosgrove D. Laminin α2-mediated focal adhesion kinase activation triggers Alport glomerular pathogenesis. PloS one. 2014:9(6):e99083. doi: 10.1371/journal.pone.0099083. Epub 2014 Jun 10 [PubMed PMID: 24915008]
Level 3 (low-level) evidenceDufek B, Meehan DT, Delimont D, Cheung L, Gratton MA, Phillips G, Song W, Liu S, Cosgrove D. Endothelin A receptor activation on mesangial cells initiates Alport glomerular disease. Kidney international. 2016 Aug:90(2):300-310. doi: 10.1016/j.kint.2016.02.018. Epub 2016 Apr 27 [PubMed PMID: 27165837]
Gunwar S, Ballester F, Noelken ME, Sado Y, Ninomiya Y, Hudson BG. Glomerular basement membrane. Identification of a novel disulfide-cross-linked network of alpha3, alpha4, and alpha5 chains of type IV collagen and its implications for the pathogenesis of Alport syndrome. The Journal of biological chemistry. 1998 Apr 10:273(15):8767-75 [PubMed PMID: 9535854]
Level 3 (low-level) evidenceCosgrove D, Rodgers K, Meehan D, Miller C, Bovard K, Gilroy A, Gardner H, Kotelianski V, Gotwals P, Amatucci A, Kalluri R. Integrin alpha1beta1 and transforming growth factor-beta1 play distinct roles in alport glomerular pathogenesis and serve as dual targets for metabolic therapy. The American journal of pathology. 2000 Nov:157(5):1649-59 [PubMed PMID: 11073824]
Level 3 (low-level) evidenceCosgrove D. Glomerular pathology in Alport syndrome: a molecular perspective. Pediatric nephrology (Berlin, Germany). 2012 Jun:27(6):885-90. doi: 10.1007/s00467-011-1868-z. Epub 2011 Apr 1 [PubMed PMID: 21455721]
Level 3 (low-level) evidenceOlaru F, Luo W, Wang XP, Ge L, Hertz JM, Kashtan CE, Sado Y, Segal Y, Hudson BG, Borza DB. Quaternary epitopes of α345(IV) collagen initiate Alport post-transplant anti-GBM nephritis. Journal of the American Society of Nephrology : JASN. 2013 May:24(6):889-95. doi: 10.1681/ASN.2012100978. Epub 2013 Apr 25 [PubMed PMID: 23620401]
Level 3 (low-level) evidenceWang XP, Fogo AB, Colon S, Giannico G, Abul-Ezz SR, Miner JH, Borza DB. Distinct epitopes for anti-glomerular basement membrane alport alloantibodies and goodpasture autoantibodies within the noncollagenous domain of alpha3(IV) collagen: a janus-faced antigen. Journal of the American Society of Nephrology : JASN. 2005 Dec:16(12):3563-71 [PubMed PMID: 16236801]
Level 3 (low-level) evidenceBorza DB. Autoepitopes and alloepitopes of type IV collagen: role in the molecular pathogenesis of anti-GBM antibody glomerulonephritis. Nephron. Experimental nephrology. 2007:106(2):e37-43 [PubMed PMID: 17570938]
Kashtan CE. Alport syndrome and thin glomerular basement membrane disease. Journal of the American Society of Nephrology : JASN. 1998 Sep:9(9):1736-50 [PubMed PMID: 9727383]
Thorner PS. Alport syndrome and thin basement membrane nephropathy. Nephron. Clinical practice. 2007:106(2):c82-8 [PubMed PMID: 17570934]
Patey-Mariaud de Serre N, Garfa M, Bessiéres B, Noël LH, Knebelmann B. Collagen alpha5 and alpha2(IV) chain coexpression: analysis of skin biopsies of Alport patients. Kidney international. 2007 Aug:72(4):512-6 [PubMed PMID: 17554254]
Gubler MC. Diagnosis of Alport syndrome without biopsy? Pediatric nephrology (Berlin, Germany). 2007 May:22(5):621-5 [PubMed PMID: 17143627]
Gubler M, Levy M, Broyer M, Naizot C, Gonzales G, Perrin D, Habib R. Alport's syndrome. A report of 58 cases and a review of the literature. The American journal of medicine. 1981 Mar:70(3):493-505 [PubMed PMID: 7211891]
Izzedine H, Tankere F, Launay-Vacher V, Deray G. Ear and kidney syndromes: molecular versus clinical approach. Kidney international. 2004 Feb:65(2):369-85 [PubMed PMID: 14717907]
Shaw EA, Colville D, Wang YY, Zhang KW, Dagher H, Fassett R, Guymer R, Savige J. Characterization of the peripheral retinopathy in X-linked and autosomal recessive Alport syndrome. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association. 2007 Jan:22(1):104-8 [PubMed PMID: 17071739]
Byrne MC, Budisavljevic MN, Fan Z, Self SE, Ploth DW. Renal transplant in patients with Alport's syndrome. American journal of kidney diseases : the official journal of the National Kidney Foundation. 2002 Apr:39(4):769-75 [PubMed PMID: 11920343]
Perrin D, Jungers P, Grünfeld JP, Delons S, Noël LH, Zenatti C. Perimacular changes in Alport's syndrome. Clinical nephrology. 1980 Apr:13(4):163-7 [PubMed PMID: 7379367]
Shah SN, Weinberg DV. Giant macular hole in Alport syndrome. Ophthalmic genetics. 2010 Jun:31(2):94-7. doi: 10.3109/13816811003767128. Epub [PubMed PMID: 20450313]
Level 3 (low-level) evidencePlevová P, Gut J, Janda J. Familial hematuria: A review. Medicina (Kaunas, Lithuania). 2017:53(1):1-10. doi: 10.1016/j.medici.2017.01.002. Epub 2017 Jan 31 [PubMed PMID: 28236514]
Cosgrove D, Liu S. Collagen IV diseases: A focus on the glomerular basement membrane in Alport syndrome. Matrix biology : journal of the International Society for Matrix Biology. 2017 Jan:57-58():45-54. doi: 10.1016/j.matbio.2016.08.005. Epub 2016 Aug 27 [PubMed PMID: 27576055]
Citirik M, Batman C, Men G, Tuncel M, Zilelioglu O. Electron microscopic examination of the anterior lens capsule in a case of Alport's syndrome. Clinical & experimental optometry. 2007 Sep:90(5):367-70 [PubMed PMID: 17697183]
Level 3 (low-level) evidenceAhmed F, Kamae KK, Jones DJ, Deangelis MM, Hageman GS, Gregory MC, Bernstein PS. Temporal macular thinning associated with X-linked Alport syndrome. JAMA ophthalmology. 2013 Jun:131(6):777-82. doi: 10.1001/jamaophthalmol.2013.1452. Epub [PubMed PMID: 23572034]
Raimundo M, Fonseca C, Silva R, Figueira J. Bilateral giant macular holes: A rare manifestation of Alport syndrome. European journal of ophthalmology. 2019 Jan:29(1):NP13-NP16. doi: 10.1177/1120672118781232. Epub 2018 Jun 6 [PubMed PMID: 29873249]
Uliana V, Marcocci E, Mucciolo M, Meloni I, Izzi C, Manno C, Bruttini M, Mari F, Scolari F, Renieri A, Salviati L. Alport syndrome and leiomyomatosis: the first deletion extending beyond COL4A6 intron 2. Pediatric nephrology (Berlin, Germany). 2011 May:26(5):717-24. doi: 10.1007/s00467-010-1693-9. Epub 2010 Dec 14 [PubMed PMID: 21380622]
Level 3 (low-level) evidenceLiapis H, Foster K, Miner JH. Red cell traverse through thin glomerular basement membrane. Kidney international. 2002 Feb:61(2):762-3 [PubMed PMID: 11849422]
Level 3 (low-level) evidenceCheong HI, Kashtan CE, Kim Y, Kleppel MM, Michael AF. Immunohistologic studies of type IV collagen in anterior lens capsules of patients with Alport syndrome. Laboratory investigation; a journal of technical methods and pathology. 1994 Apr:70(4):553-7 [PubMed PMID: 8176894]
Gyoneva L, Segal Y, Dorfman KD, Barocas VH. Mechanical response of wild-type and Alport murine lens capsules during osmotic swelling. Experimental eye research. 2013 Aug:113():87-91. doi: 10.1016/j.exer.2013.05.008. Epub 2013 May 21 [PubMed PMID: 23707242]
Level 3 (low-level) evidenceSavige J, Ariani F, Mari F, Bruttini M, Renieri A, Gross O, Deltas C, Flinter F, Ding J, Gale DP, Nagel M, Yau M, Shagam L, Torra R, Ars E, Hoefele J, Garosi G, Storey H. Expert consensus guidelines for the genetic diagnosis of Alport syndrome. Pediatric nephrology (Berlin, Germany). 2019 Jul:34(7):1175-1189. doi: 10.1007/s00467-018-3985-4. Epub 2018 Jul 9 [PubMed PMID: 29987460]
Level 3 (low-level) evidenceRumpelt HJ. Hereditary nephropathy (Alport syndrome): correlation of clinical data with glomerular basement membrane alterations. Clinical nephrology. 1980 May:13(5):203-7 [PubMed PMID: 7398144]
Gettelfinger JD, Dahl JP. Syndromic Hearing Loss: A Brief Review of Common Presentations and Genetics. Journal of pediatric genetics. 2018 Mar:7(1):1-8. doi: 10.1055/s-0037-1617454. Epub 2018 Jan 4 [PubMed PMID: 29441214]
Easson A, Walter S. Hearing-impaired young people - a physician's guide . Clinical medicine (London, England). 2017 Dec:17(6):521-524. doi: 10.7861/clinmedicine.17-6-521. Epub [PubMed PMID: 29196352]
Gross O, Licht C, Anders HJ, Hoppe B, Beck B, Tönshoff B, Höcker B, Wygoda S, Ehrich JH, Pape L, Konrad M, Rascher W, Dötsch J, Müller-Wiefel DE, Hoyer P, Study Group Members of the Gesellschaft für Pädiatrische Nephrologie, Knebelmann B, Pirson Y, Grunfeld JP, Niaudet P, Cochat P, Heidet L, Lebbah S, Torra R, Friede T, Lange K, Müller GA, Weber M. Early angiotensin-converting enzyme inhibition in Alport syndrome delays renal failure and improves life expectancy. Kidney international. 2012 Mar:81(5):494-501. doi: 10.1038/ki.2011.407. Epub 2011 Dec 14 [PubMed PMID: 22166847]
Level 2 (mid-level) evidenceDaina E, Cravedi P, Alpa M, Roccatello D, Gamba S, Perna A, Gaspari F, Remuzzi G, Ruggenenti P. A multidrug, antiproteinuric approach to alport syndrome: a ten-year cohort study. Nephron. 2015:130(1):13-20. doi: 10.1159/000381480. Epub 2015 Apr 21 [PubMed PMID: 25895746]
Gross O, Tönshoff B, Weber LT, Pape L, Latta K, Fehrenbach H, Lange-Sperandio B, Zappel H, Hoyer P, Staude H, König S, John U, Gellermann J, Hoppe B, Galiano M, Hoecker B, Ehren R, Lerch C, Kashtan CE, Harden M, Boeckhaus J, Friede T, German Pediatric Nephrology (GPN) Study Group and EARLY PRO-TECT Alport Investigators. A multicenter, randomized, placebo-controlled, double-blind phase 3 trial with open-arm comparison indicates safety and efficacy of nephroprotective therapy with ramipril in children with Alport's syndrome. Kidney international. 2020 Jun:97(6):1275-1286. doi: 10.1016/j.kint.2019.12.015. Epub 2020 Jan 17 [PubMed PMID: 32299679]
Level 1 (high-level) evidenceNicklason E, Mack H, Beltz J, Jacob J, Farahani M, Colville D, Savige J. Corneal endothelial cell abnormalities in X-linked Alport syndrome. Ophthalmic genetics. 2020 Feb:41(1):13-19. doi: 10.1080/13816810.2019.1709126. Epub 2020 Mar 11 [PubMed PMID: 32159412]
Antignac C, Knebelmann B, Drouot L, Gros F, Deschênes G, Hors-Cayla MC, Zhou J, Tryggvason K, Grünfeld JP, Broyer M. Deletions in the COL4A5 collagen gene in X-linked Alport syndrome. Characterization of the pathological transcripts in nonrenal cells and correlation with disease expression. The Journal of clinical investigation. 1994 Mar:93(3):1195-207 [PubMed PMID: 8132760]
Hudson BG, Kalluri R, Gunwar S, Weber M, Ballester F, Hudson JK, Noelken ME, Sarras M, Richardson WR, Saus J. The pathogenesis of Alport syndrome involves type IV collagen molecules containing the alpha 3(IV) chain: evidence from anti-GBM nephritis after renal transplantation. Kidney international. 1992 Jul:42(1):179-87 [PubMed PMID: 1635348]
Level 3 (low-level) evidenceBrainwood D, Kashtan C, Gubler MC, Turner AN. Targets of alloantibodies in Alport anti-glomerular basement membrane disease after renal transplantation. Kidney international. 1998 Mar:53(3):762-6 [PubMed PMID: 9507224]
Kalluri R, van den Heuvel LP, Smeets HJ, Schroder CH, Lemmink HH, Boutaud A, Neilson EG, Hudson BG. A COL4A3 gene mutation and post-transplant anti-alpha 3(IV) collagen alloantibodies in Alport syndrome. Kidney international. 1995 Apr:47(4):1199-204 [PubMed PMID: 7783419]
Tryggvason K, Zhou J, Hostikka SL, Shows TB. Molecular genetics of Alport syndrome. Kidney international. 1993 Jan:43(1):38-44 [PubMed PMID: 8433568]
Kashtan CE, Michael AF. Alport syndrome. Kidney international. 1996 Nov:50(5):1445-63 [PubMed PMID: 8914010]
Quérin S, Noël LH, Grünfeld JP, Droz D, Mahieu P, Berger J, Kreis H. Linear glomerular IgG fixation in renal allografts: incidence and significance in Alport's syndrome. Clinical nephrology. 1986 Mar:25(3):134-40 [PubMed PMID: 3514017]
Kashtan CE, Butkowski RJ, Kleppel MM, First MR, Michael AF. Posttransplant anti-glomerular basement membrane nephritis in related males with Alport syndrome. The Journal of laboratory and clinical medicine. 1990 Oct:116(4):508-15 [PubMed PMID: 2212860]
Level 3 (low-level) evidenceKajimoto Y, Endo Y, Terasaki M, Kunugi S, Igarashi T, Mii A, Terasaki Y, Shimizu A. Pathologic glomerular characteristics and glomerular basement membrane alterations in biopsy-proven thin basement membrane nephropathy. Clinical and experimental nephrology. 2019 May:23(5):638-649. doi: 10.1007/s10157-018-01687-1. Epub 2019 Jan 28 [PubMed PMID: 30687875]
Massella L, Gangemi C, Giannakakis K, Crisafi A, Faraggiana T, Fallerini C, Renieri A, Muda AO, Emma F. Prognostic value of glomerular collagen IV immunofluorescence studies in male patients with X-linked Alport syndrome. Clinical journal of the American Society of Nephrology : CJASN. 2013 May:8(5):749-55. doi: 10.2215/CJN.07510712. Epub 2013 Jan 31 [PubMed PMID: 23371956]
Level 2 (mid-level) evidenceJais JP, Knebelmann B, Giatras I, De Marchi M, Rizzoni G, Renieri A, Weber M, Gross O, Netzer KO, Flinter F, Pirson Y, Dahan K, Wieslander J, Persson U, Tryggvason K, Martin P, Hertz JM, Schröder C, Sanak M, Carvalho MF, Saus J, Antignac C, Smeets H, Gubler MC. X-linked Alport syndrome: natural history and genotype-phenotype correlations in girls and women belonging to 195 families: a "European Community Alport Syndrome Concerted Action" study. Journal of the American Society of Nephrology : JASN. 2003 Oct:14(10):2603-10 [PubMed PMID: 14514738]
Rheault MN. Women and Alport syndrome. Pediatric nephrology (Berlin, Germany). 2012 Jan:27(1):41-6. doi: 10.1007/s00467-011-1836-7. Epub 2011 Mar 5 [PubMed PMID: 21380623]
Level 3 (low-level) evidenceFallerini C, Baldassarri M, Trevisson E, Morbidoni V, La Manna A, Lazzarin R, Pasini A, Barbano G, Pinciaroli AR, Garosi G, Frullanti E, Pinto AM, Mencarelli MA, Mari F, Renieri A, Ariani F. Alport syndrome: impact of digenic inheritance in patients management. Clinical genetics. 2017 Jul:92(1):34-44. doi: 10.1111/cge.12919. Epub 2017 Feb 22 [PubMed PMID: 27859054]