Introduction
Bernard-Soulier syndrome (BSS), also known as Hemorrhagiparous Thrombolytic Dystrophy, is an extremely rare inherited blood clotting disorder characterized by giant platelet cells, thrombocytopenia, and prolonged bleeding time. In 1948, Jean-Bernard and Jean-Pierre Soulier described the first male patient who presented with repeated episodes of bleeding throughout his life and eventually died at the age of 28 years from an intracranial hemorrhage sustained after a bar fight.[1][2] BSS involves a defect of the GPIb-IX-V complex, an essential platelet receptor complex that principally binds with the von Willebrand factor (vWF). However, it has multiple other functions in inducing thrombosis and hemostasis.[3]
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
BSS is the result of genetic mutations encoding for GPIb-alpha (GPIBA), GPIB-beta (GPIBB), and GPIX (GP9), which are 3 of the 4 subunits that make the GPIb-IX-V complex.[4] Nearly 112 mutations were identified in a study of 211 families with BSS. Many variations were homozygotes and usually the product of consanguinous marriages.[5] A significant number of families tested have proven to have asymptomatic carriers. These types are inherited as autosomal dominant; the most notable of these is exemplified by the Bolzano mutation (c.515C>T).[6] These mutations were mainly identified in the GP1BA (28%), GP1BB (28%), or GP9 (44%) genes.[4] These heterogeneous mutations could be nonsense, missense, frameshift, deletion, or insertion.[7] Most of these mutations are inherited in an autosomal recessive pattern; however, rare cases of autosomal dominant inheritance have been reported.[4]
Patients with mutations in both alleles (autosomal recessive inheritance) are referred to as having biallelic BSS (bBSS), and those with a mutation in only a single allele (autosomal dominant inheritance) are often referred to as monoallelic BSS (mBSS).[2] The biallelic "loss-of-function" variants (with deletions, insertions, and nonsense mutations) in GP1BA, GP1BB, or GP9 cause disruptions in the GP1b/IX/V complex, often preventing its formation.[5][8] This complex is the receptor for vWF and thrombin. Other variants involving GP targeting and inheritance can be found worldwide, including China (c.97T>A), Denmark (c.58T>G), Reunion (French) Isle (c.265A>G), and the Czech Republic (pC33Y).[9][10][11][12]
Epidemiology
The prevalence of the disease is estimated to be 1 per million. However, the prevalence may be much higher than estimated due to under-recognition and misdiagnosis.[4][13] BSS is inherited via autosomal genes, hence, it is prevalent in both male and female patients.[2][14]
As the disease is widely underrecognized, it is not often diagnosed in the early stages of life, and the average age of diagnosis is 16 years. Many patients are diagnosed with idiopathic thrombocytopenic purpura (ITP) and even go on to receive a splenectomy. Thus, it is imperative to understand the presenting features and pathophysiology of BSS to correctly diagnose the disease.[4]
Pathophysiology
The GPIb-IX-V complex is expressed on the platelet surface. By facilitating platelet adhesion to the subendothelium, the GPIb-IX-V complex leads to clot formation whenever the vascular subendothelium is exposed or a plaque ruptures. The activity of GPIb-IX-V complex is also critical in deep venous thrombosis (DVT).[3]
The most important function of the GPIb-IX-V complex is to bind with VWF and initiate a signaling cascade that activates the platelet integrin GPIIb-IIIa leading to platelet aggregation. Although VWF is also a weak agonist, a full activation signal is required via the thromboxane A2 and ADP-dependent signaling pathway to activate the platelets.[3][15]
The N-terminal of GPIb-Alpha plays a critical role in platelet-mediated coagulation by providing binding sites to high molecular weight (HMW) kininogen, Factors XI, XII, and alpha-thrombin. The same N-terminal of GPIb-alpha is a primary binding site for multiple ligands. It serves as a pivotal point for the cross-talk between platelets and leukocytes in thrombosis and inflammatory response.[3][15]
The GPIb-IX-V also plays a role in maintaining platelet shape by linking the platelet surface to a sub-membranous network of actin filaments, the platelet membrane skeleton. This involves the central portion of the cytoplasmic tail of GPIb-alpha, particularly Phe568 and Trp570, which provides a binding site for the actin-associated protein, filamin A.[3]
Considering all the functions of the GPIb-IX-V complex, one can understand that a mutation in the encoding genes can lead to reduced activation of the platelets, defective adhesion, and, subsequently, inadequate clot-forming capability. In addition to this, the defects in the complex can also explain the giant platelets in patients with BSS.
History and Physical
The typical presentation of BSS starts at birth and continues throughout life. It is characterized by bleeding from different sites, epistaxis, cutaneous bleeding, hemorrhage after trauma, eg, brain hemorrhage after head trauma, prolonged bleeding after dental procedures, and heavy menstrual bleeding in females.[15][14] More rarely reported symptoms are gastrointestinal bleeding and hematuria. Clinical features can be limited to unexplained purpura or bruising only. On the contrary, bleeding can be fatal in about 16% of reported cases.[2][1] Spontaneous intracranial hemorrhage or intraarticular hemorrhages are not common. Fatalities from BSS are very rare.[2]
The patients with BSS who present in adulthood are usually those with mBSS. Such patients have less number of bleeding episodes due to preserved platelet numbers.[2][16] A significant monoallelic mutation is the 'Ala156Val' mutation in the GPIB-alpha, called the Bolzano mutation. Although most patients will only have mild thrombocytopenia and infrequent bleeding episodes, a few patients have been described in whom the bleeding was very severe.[16] Similarly, other mutations have also been described where only mild thrombocytopenia was noted with mild bleeding episodes.[2]
The International Society on Thrombosis and Haemostasis Bleeding Assessment Tool (ISTH-BAT) is useful for assessing bleeding disorders. Its utility was tested in a small study, including patients with known inherited platelet disorders. The study demonstrated a specificity of 100%, a positive predictive value of 90%, and a negative predictive value of 100% using this assessment tool.[17] In the setting of von Willebrand disease, a BAT score >6 repeatedly correlates to a 99% probability of an inherited platelet defect such as BSS.[8]] Similarly, other bleeding assessment tools like Molecular and Clinical Markers for the Diagnosis and Management (MCMDM) of type 1 von Willebrand's disease (VWD) and the World Health Organization Bleeding Assessment Tool are similar tools. An electronic version of MCMDM-type-1 VWD was developed in 2010.[17]
Evaluation
BSS should be considered in the differential diagnosis of any patient who presents with a prolonged bleeding history, especially if the bleeding history started from early childhood.
Most patients with BSS have a platelet count between 20 to 100 billion/L. However, a count as low as 10 x 10^9/L has been reported.[15] The peripheral smear usually presents thrombocytopenia and large platelets (see Image. Bernard Soulier Syndrome). If there is a debate on whether the platelet population is enlarged, one can check the mean platelet volume (MPV) from the Hematologic Analyzer. An MPV >12.4 fl has a high likelihood of enlargement.[8] The bleeding time is significantly prolonged.[18] The platelet function analyzer (PFA-100) closure time is prolonged, usually in the adenosine-diphosphate (ADP) and epinephrine cartridges.[18]
Platelet aggregation studies (also called light transmission aggregometry) demonstrate a reduced response to ristocetin that is not corrected by the addition of normal plasma.[19] This feature helps to distinguish BSS from VWD. The responses to ADP, collagen, and arachidonic acid are normal. However, in a few patients, platelet aggregation in response to thrombin is reduced.[14]
Flow cytometry of platelet glycoprotein is the confirmatory test. It demonstrates a marked reduction of CD42a (GPIX) and CD42b (GPIb-alpha). Since flow cytometry requires only small volumes of blood, this is an appropriate test for neonates, infants, and young children.[20] Molecular genetics can identify genetic abnormalities and identify affected family members as well.
In patients with mBSS, specifically in those with Bolzano mutation (the most frequent cause of inherited thrombocytopenia in Italy) or other similar mutations, the GPIb-IX-V complex is present in normal numbers, albeit defective, and cannot bind to vWF. In such patients, the near-absent ristocetin-induced platelet aggregation is a good diagnostic tool.[3]
Treatment / Management
Preventive Care
- Patients diagnosed with BSS should be educated extensively about the risks of bleeding. They should carry 'alert cards' or wear 'alert bracelets' identifying the diagnosis of BSS. They should be registered with a center that can provide emergent treatment 24 hours a day should they present with a bleeding episode. The patients should also be educated on maintaining dental hygiene, avoiding high-risk sports (especially contact sports), and applying pressure for epistaxis.[2]
- Patients, their family members, and their providers should be advised extensively about which medications can increase the risk of bleeding (anti-histamines, nonsteroidal anti-inflammatory drugs, and certain antibiotics). Similarly, foods, beverages, and herbal medicines that can affect the platelet function or number must be avoided.
- All patients with BSS, especially women, can suffer from iron deficiency due to excessive bleeding and should be monitored closely for the same and supplemented with iron if needed.[2]
- HLA-typing should be done for each patient at the time of diagnosis. All attempts should be made to procure HLA-matched platelets before transfusion.
Treatment Directed Towards Bleeding Episodes
- Platelet transfusions - These are the first line of treatment in patients with BSS who present with acute hemorrhage or are preparing for elective surgery. However, with each transfusion comes the risk of alloantibody formation and a minuscule risk of transmission of pathogens.
- Transmission of bacteria - As platelets are stored at a higher temperature (20 °C to 24 °C), there is always a risk of bacterial growth and transmission. The risk is much higher with whole-blood-derived platelets compared to apheresis-derived platelets.[21]
- Alloimmunization - HLA-matched platelets are ideal for transfusion but may not be available in emergent conditions.[2] Alloimmunization against human platelet antigen or human leukocyte antigen is common for patients requiring frequent transfusions and can be associated with platelet refractoriness and platelet transfusion failure. A Brazilian center developed an algorithm for evaluating the presence of platelet alloantibodies; however, its tests have shortcomings. Platelet immunofluorescence lacks specificity, while protocols with monoclonal antibody immobilization of platelet antigens (MAIPA) and bead-based Luminex platelet assay are labor-intensive and costly.[22]
- Antibodies against glycoproteins - Although this is a more common phenomenon in patients with Glanzmann's thrombasthenia (GT), patients with BSS can also develop antibodies against glycoproteins that they lack.[14]
- Antifibrinolytic therapy - Tranexamic acid has been used successfully in managing mucocutaneous bleeding and menorrhagia in conjunction with local efforts like nasal packing, compression sponges, hormonal treatment, etc.[19] However, the use of antifibrinolytics should be avoided in patients with pulmonary hemorrhage or those with active hematuria due to the risk of the formation of intraluminal clots leading to respiratory failure and renal failure, respectively.
- Desmopressin (DDAVP) - Releases vWF from the endothelial cells. Due to a defective GPIB-IX-V complex, the utility of DDAVP in managing BSS is severely limited. Only anecdotal reports suggest the use of DDAVP. Clinicians must be aware of the potential adverse effects of severe hyponatremia and seizures associated with repeated DDAVP use.[23]
Potentially Beneficial Strategies NOT Approved for Patients With BSS
- In a small phase II clinical trial, the thrombopoietin receptor agonist, Eltrombopag, was successfully used to increase the platelet count in patients diagnosed with inherited thrombocytopenias. One of the 5 patients had monoallelic BSS. Eltrombopag is not approved for treating thrombocytopenia secondary to BSS.[24] In a different study, Eltrombopag was found to be efficacious in many forms of inherited thrombocytopenia, including BSS.[24] Positive responses theoretically support using thrombopoietin in the perioperative setting to prevent alloimmunization and its subsequent refractoriness to platelet transfusions.
- Recombinant factor VII (rfVII) has been approved for use in patients with Glanzmann's thrombasthenia (GT) but not in patients with BSS. There are reports of successful use of rfVII in patients with BSS. The United Kingdom Haemophilia Centre Doctors' Organisation (UKHCDO) 2006 guidelines and the British Society of Haematology (BSH) platelet transfusion guidelines both recommend the use of recombinant factor VII (rfVII) in patients with GT or BSS in the event of severe bleeding.[18][25] Other studies have reported the use of rFVIIr in the treatment of severe bleeding episodes in BSS where cases were refractory to platelet transfusions.[26][27][22]
- Allogeneic stem cell transplant has been used in patients with BSS. However, there is scarce data.[28][29] This is usually reserved for patients with too many antibodies and with a severe bleeding disorder. (A1)
Special Considerations in Pregnant Patients With BSS [2]
- Counseling prospective parents: Identify potential risk factors (consanguineous marriage, parents are carriers, etc) for the fetus to develop BSS. In women with BSS, the potential risk of hemorrhage should be discussed. Neonates will not develop homozygous BSS unless both parents are carriers.
- Antenatal: Manage specialized units in consultation with high-risk obstetrics and hematology. The mother should be checked for HLA type and anti-platelet antibodies and assessed for the risk of developing neonatal alloimmune thrombocytopenia (FNAIT).
- Labor: Neuraxial anesthesia is contraindicated as hemostasis cannot be guaranteed. The use of uterotonics is encouraged in the second stage of labor. HLA-matched platelets and tranexamic acid should be used if needed. The rfVII can be used in severe bleeding.
- Postpartum: All patients must be monitored for a minimum of 8 weeks for bleeding symptoms.
- Neonates: The risk of FNAIT is high, especially in mothers diagnosed with BSS requiring multiple platelet transfusions throughout their lives. Such patients have anti-GPI antibodies that can cross the placenta and affect the normal platelets of the neonate. The blood count of neonates should be monitored very closely.
Differential Diagnosis
The differential diagnosis for BSS starts by considering various bleeding disorders, including factor deficiencies. Generally, the patient's presentation may guide one in the correct diagnostic direction. Mucocutaneous bleeding, coupled with large platelets and thrombocytopenia, points towards a platelet disorder. Large platelets or thrombocytopenia on peripheral smear review is NOT a feature of hemophilia.
Immune Thrombocytopenia
Many patients with BSS are diagnosed with immune thrombocytopenia due to similar presentations. BSS should always be considered in the differential of a persistent or refractory ITP.[30] A reevaluation with aggregation studies (especially ristocetin) and flow cytometry will be needed to confirm the diagnosis of BSS. Features that may help in differentiating BSS from ITP are the following:
- ITP is typically an acquired disease and is not likely to have a familial pattern. BSS is autosomal recessive in large part and common in countries with a high consanguineous marriage rate.
- Failure to respond to first-line treatment like intravenous immunoglobulin and steroids
- Typical findings for BSS on aggregation studies and flow cytometry. In the former, there is an absence of aggregation to Risgtocetin, while the others are normal. Flow studies of GP1b (CD42) and GPIX (CD42a) are important as the defective or absent expression of GP1b/IX is characteristic of BSS.[30]
Despite this, many patients mistakenly diagnosed with ITP end up receiving splenectomy before being diagnosed with BSS.[2]
Von Willebrand Disease (vWD)
The patients with type IIB vWD have the closest clinical phenotype with BSS. Due to the increased affinity of large multimers with platelets, the platelets are cleared rapidly, leading to thrombocytopenia. The platelets are also large in type IIB vWD. However, the platelets express an increased aggregation in response to ristocetin. In comparison, patients with BSS always have low to absent aggregation of platelets in response to ristocetin.[31] Also, patients with platelet-type vWD carry a mutation in the GP1b-alpha. However, this mutation increases the affinity of platelets to vWF.[32]
Other Inherited Disorders in the Differential Diagnosis of BSS
- May-Hegglin abnormality
- Myosin-Heavy chain 9 (MYH-9) disorders
- Grey Platelet syndrome- lack of intra-platelet granules confirmed by electron microscopy.[33]
- Paris Trousseau Platelet disorder (PTPD) - deletion of the terminal end of the long arm of chromosome 11 that includes band 11q24.1, 11q terminal deletion disorder.[34]
- DiGeorge syndrome (DGS), velocardiofacial syndrome (VCFS, or Shprintzen syndrome), and conotruncal anomaly of the face share the microdeletion of chromosome 22q11.2.[35] Patients usually are heterozygotes for BSS.[36]
- Mediterranean macrothrombocytopenia [35][37]
Rare cases of autoantibody to GPIb complex have been described, called Pseudo-BSS.[38][39]
Prognosis
If BSS patients receive good primary care and education about avoiding trauma and following precautions, they can live their lives quite normally. Preventive care and meticulous planning for elective surgeries are needed to minimize bleeding events. Patients must always wear alert bracelets to alert emergency services in case of trauma.[1]
Complications
Patients with BSS frequently suffer from bleeding complications due to the nature of the disease. Other complications include:
- Transmission of bloodborne pathogens - The risk though is minimal due to extensive screening of blood products. Still, bacterial transmission can occur due to platelets being stored at a higher temperature.
- Development of autoantibodies due to repeated blood transfusions- While alloimmunization can be sidestepped with HLA-matched products, this may not be an option in urgent or emergent settings.
- Iron deficiency anemia, especially in women who suffer from menorrhagia, results in constant fatigue and loss of work hours.
- Transmission of antibodies across the placenta may lead to fetal/neonatal alloimmune thrombocytopenia.
- Concurrent ailments can obscure the presence of BSS and may only be discovered serendipitously or catastrophically. Recent work has told of a patient with multiple myeloma who developed an underlying acquired BSS.[40] Another situation was reported when a patient underwent a thyroidectomy only to develop severe postoperative bleeding.[41] Hemostasis was achieved after repeated transfusions of platelets (along with packed cells and plasma) were given. Preoperatively, the patient had manifested only a mild decrease in the platelet count; postoperatively, he manifested macrothrombocytopenia. Postoperative analysis revealed his BSS. While some might see BSS as low on the diagnostic list, its obscurity and bleeding threat warrant a higher consideration.
- In patients with BSS and coronary artery disease (CAD), percutaneous coronary intervention (PCI) has warranted special attention.[42] To protect patients against procedural hemorrhage, they should receive preoperative platelet transfusions. It has been advocated to reduce dual-antiplatelet therapy (DAPT) to single-antiplatelet therapy. Preprocedural platelet transfusions are followed by drug-eluting stents, and postprocedural DAPT is supported.
Consultations
All healthcare professionals caring for patients diagnosed with BSS must emphasize preventive care to patients and families. In addition, the following consultants must be involved.
- A hematologist (pediatric or adult) must be involved in case the patient requires emergency or elective surgery.
- In the event of planning for pregnancy, high-risk obstetrics and gynecology teams must be involved.
- Transfusion medicine must be consulted whenever blood products are required to meticulously screen for antibodies and provide for HLA-matched platelets.
Deterrence and Patient Education
BSS syndrome is a rare inherited bleeding disorder. It is most commonly misdiagnosed as ITP, which leads to unnecessary interventions like splenectomy. The most common presentation is that of a bleeding child, presenting with thrombocytopenia and giant platelets. The most common differential diagnosis is vWD, especially type IIB vWD and platelet-type vWD, which have a similar presentation. Low to complete absence of platelet aggregation in response to ristocetin strongly points towards a diagnosis of BSS. Flow cytometry usually clinches the diagnosis of BSS.
Preventative measures are the best for preventing bleeding complications from BSS. Patients diagnosed with BSS and their families must be educated about the nature of the disease and the potential bleeding complications. They must be taught about the environmental (contact sports, trauma, using a soft toothbrush, etc) and medicinal (nonsteroidal anti-inflammatory drugs, aspirin, etc) factors that can increase the risk of bleeding and how to avoid them. The patients and their families should be educated on techniques to stop bleeding (pressure application for epistaxis, gum bleeding, etc).
Healthcare teams managing patients diagnosed with BSS must plan ahead of time to mitigate bleeding. HLA-matched platelets must be made available for patients requiring elective surgeries. All patients must be registered in a hospital that can provide 24-hour emergency care in the advent of uncontrolled bleeding.
Pearls and Other Issues
Key facts to keep in mind regarding BSS are as follows:
- Maintain a high index of suspicion for BSS in a bleeding patient with large platelets and thrombocytopenia, especially if they do not respond to the first-line treatment for ITP.
- Low to absent response to ristocetin is a reliable indicator for diagnosing BSS (rules out vWD). Flow cytometry is used to confirm the diagnosis.
- Platelet transfusion is the first line of treatment for a bleeding patient. Antifibrinolytics can be used to control mucocutaneous bleeding.
- Desmopressin has no role in the treatment of BSS.
- Activated rfVII is not approved for BSS but has been successfully used in anecdotal cases.
- HLA-matched platelets must be used in all planned surgeries.
- Prevention of bleeding is the best approach in patients diagnosed with BSS. Extensive education programs must be conducted for patients and families with BSS.
Enhancing Healthcare Team Outcomes
In managing BSS, healthcare professionals, including physicians, advanced care practitioners, nurses, pharmacists, and other team members, must employ a multifaceted approach to ensure patient-centered care, optimize outcomes, prioritize patient safety, and enhance overall team performance.
Professionals involved in BSS care must possess specialized skills to diagnose this rare clotting disorder. The diagnosis of BSS requires a high index of suspicion, skills in differentiating BSS from other platelet disorders, and expertise in interpreting genetic data. A diagnosis of BSS must be considered in any bleeding patient with thrombocytopenia and large platelets, especially if they present in childhood.
Developing a comprehensive strategy for affected patients involves creating tailored treatment plans that consider the severity of BSS, patient age, and comorbidities. A strategic approach should include regular assessments, preventive measures, and a focus on patient education to empower individuals and their families to manage the condition effectively.
Coordinating care for individuals with BSS requires a patient-centric and interprofessional approach. All patients with BSS must be registered in centers that can provide 24-hour, tertiary-level care in the event of bleeding. Professionals should collaborate on treatment plans, share insights on patient progress, and collectively address emerging challenges. Coordinated care ensures a continuum of support, minimizes gaps in the care process, and optimizes outcomes for patients and their families. This holistic and multidisciplinary approach ensures that patient safety is upheld and overall team performance is enhanced in the face of this rare and complex hematologic condition.
Media
(Click Image to Enlarge)
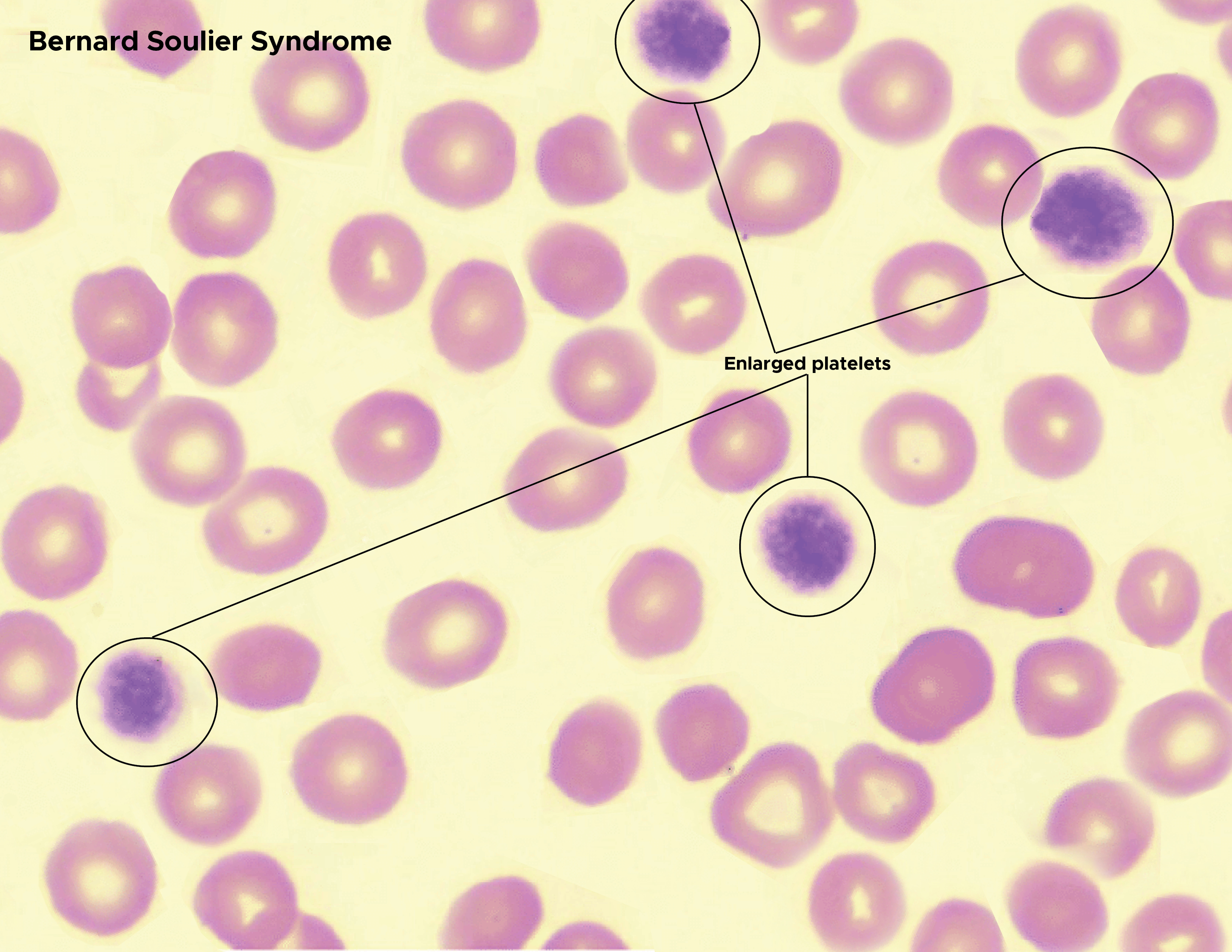
Bernard Soulier Syndrome. Enlarged platelets, red blood cells Contributed by Chelsea Rowe
References
Lanza F. Bernard-Soulier syndrome (hemorrhagiparous thrombocytic dystrophy). Orphanet journal of rare diseases. 2006 Nov 16:1():46 [PubMed PMID: 17109744]
Grainger JD, Thachil J, Will AM. How we treat the platelet glycoprotein defects; Glanzmann thrombasthenia and Bernard Soulier syndrome in children and adults. British journal of haematology. 2018 Sep:182(5):621-632. doi: 10.1111/bjh.15409. Epub 2018 Aug 17 [PubMed PMID: 30117143]
Berndt MC, Andrews RK. Bernard-Soulier syndrome. Haematologica. 2011 Mar:96(3):355-9. doi: 10.3324/haematol.2010.039883. Epub [PubMed PMID: 21357716]
Savoia A, Kunishima S, De Rocco D, Zieger B, Rand ML, Pujol-Moix N, Caliskan U, Tokgoz H, Pecci A, Noris P, Srivastava A, Ward C, Morel-Kopp MC, Alessi MC, Bellucci S, Beurrier P, de Maistre E, Favier R, Hézard N, Hurtaud-Roux MF, Latger-Cannard V, Lavenu-Bombled C, Proulle V, Meunier S, Négrier C, Nurden A, Randrianaivo H, Fabris F, Platokouki H, Rosenberg N, HadjKacem B, Heller PG, Karimi M, Balduini CL, Pastore A, Lanza F. Spectrum of the mutations in Bernard-Soulier syndrome. Human mutation. 2014 Sep:35(9):1033-45. doi: 10.1002/humu.22607. Epub 2014 Jul 15 [PubMed PMID: 24934643]
Ghalloussi D, Rousset-Rouvière C, Popovici C, Garaix F, Saut N, Saultier P, Tsimaratos M, Chambost H, Alessi MC, Baccini V. Bernard-Soulier syndrome: first human case due to a homozygous deletion of GP9 gene. British journal of haematology. 2020 Mar:188(6):e87-e90. doi: 10.1111/bjh.16374. Epub 2020 Feb 6 [PubMed PMID: 32030720]
Level 3 (low-level) evidenceMinkov M, Zeitlhofer P, Zoubek A, Kager L, Panzer S, Haas OA. Novel Compound Heterozygous Mutations in Two Families With Bernard-Soulier Syndrome. Frontiers in pediatrics. 2020:8():589812. doi: 10.3389/fped.2020.589812. Epub 2021 Jan 22 [PubMed PMID: 33553065]
Diz-Küçükkaya R. Inherited platelet disorders including Glanzmann thrombasthenia and Bernard-Soulier syndrome. Hematology. American Society of Hematology. Education Program. 2013:2013():268-75. doi: 10.1182/asheducation-2013.1.268. Epub [PubMed PMID: 24319190]
Kim B. Diagnostic workup of inherited platelet disorders. Blood research. 2022 Apr 30:57(S1):11-19. doi: 10.5045/br.2022.2021223. Epub [PubMed PMID: 35483920]
Skalníková M, Staňo Kozubík K, Trizuljak J, Vrzalová Z, Radová L, Réblová K, Holbová R, Kurucová T, Svozilová H, Štika J, Blaháková I, Dvořáčková B, Prudková M, Stehlíková O, Šmída M, Křen L, Smejkal P, Pospíšilová Š, Doubek M. A GP1BA Variant in a Czech Family with Monoallelic Bernard-Soulier Syndrome. International journal of molecular sciences. 2022 Jan 14:23(2):. doi: 10.3390/ijms23020885. Epub 2022 Jan 14 [PubMed PMID: 35055070]
Ma J, Chen Z, Li G, Gu H, Wu R. A novel mutation in GP1BA gene in a family with autosomal dominant Bernard Soulier syndrome variant: A case report. Experimental and therapeutic medicine. 2021 Apr:21(4):360. doi: 10.3892/etm.2021.9791. Epub 2021 Feb 13 [PubMed PMID: 33732333]
Level 3 (low-level) evidenceLeinøe E, Brøns N, Rasmussen AØ, Gabrielaite M, Zaninetti C, Palankar R, Zetterberg E, Rosthøj S, Ostrowski SR, Rossing M. The Copenhagen founder variant GP1BA c.58T}G is the most frequent cause of inherited thrombocytopenia in Denmark. Journal of thrombosis and haemostasis : JTH. 2021 Nov:19(11):2884-2892. doi: 10.1111/jth.15479. Epub 2021 Aug 11 [PubMed PMID: 34333846]
Fiore M, De Thoré C, Randrianaivo-Ranjatoelina H, Baas MJ, Jacquemont ML, Dreyfus M, Lavenu-Bombled C, Li R, Gachet C, Dupuis A, Lanza F. High prevalence of the natural Asn89Asp mutation in the GP1BB gene associated with Bernard-Soulier syndrome in French patients from the genetic isolate of Reunion Island. British journal of haematology. 2020 May:189(3):e67-e71. doi: 10.1111/bjh.16479. Epub 2020 Jan 30 [PubMed PMID: 31997307]
Balduini CL, Savoia A, Seri M. Inherited thrombocytopenias frequently diagnosed in adults. Journal of thrombosis and haemostasis : JTH. 2013 Jun:11(6):1006-19. doi: 10.1111/jth.12196. Epub [PubMed PMID: 23510089]
Alamelu J, Liesner R. Modern management of severe platelet function disorders. British journal of haematology. 2010 Jun:149(6):813-23. doi: 10.1111/j.1365-2141.2010.08191.x. Epub 2010 Apr 29 [PubMed PMID: 20456364]
López JA, Andrews RK, Afshar-Kharghan V, Berndt MC. Bernard-Soulier syndrome. Blood. 1998 Jun 15:91(12):4397-418 [PubMed PMID: 9616133]
Noris P, Perrotta S, Bottega R, Pecci A, Melazzini F, Civaschi E, Russo S, Magrin S, Loffredo G, Di Salvo V, Russo G, Casale M, De Rocco D, Grignani C, Cattaneo M, Baronci C, Dragani A, Albano V, Jankovic M, Scianguetta S, Savoia A, Balduini CL. Clinical and laboratory features of 103 patients from 42 Italian families with inherited thrombocytopenia derived from the monoallelic Ala156Val mutation of GPIbα (Bolzano mutation). Haematologica. 2012 Jan:97(1):82-8. doi: 10.3324/haematol.2011.050682. Epub 2011 Sep 20 [PubMed PMID: 21933849]
Kaur H, Borhany M, Azzam H, Costa-Lima C, Ozelo M, Othman M. The utility of International Society on Thrombosis and Haemostasis-Bleeding Assessment Tool and other bleeding questionnaires in assessing the bleeding phenotype in two platelet function defects. Blood coagulation & fibrinolysis : an international journal in haemostasis and thrombosis. 2016 Jul:27(5):589-93. doi: 10.1097/MBC.0000000000000496. Epub [PubMed PMID: 27100304]
Bolton-Maggs PH, Chalmers EA, Collins PW, Harrison P, Kitchen S, Liesner RJ, Minford A, Mumford AD, Parapia LA, Perry DJ, Watson SP, Wilde JT, Williams MD, UKHCDO. A review of inherited platelet disorders with guidelines for their management on behalf of the UKHCDO. British journal of haematology. 2006 Dec:135(5):603-33 [PubMed PMID: 17107346]
Mohan G, Malayala SV, Mehta P, Balla M. A Comprehensive Review of Congenital Platelet Disorders, Thrombocytopenias and Thrombocytopathies. Cureus. 2020 Oct 31:12(10):e11275. doi: 10.7759/cureus.11275. Epub 2020 Oct 31 [PubMed PMID: 33274150]
Cohn RJ, Sherman GG, Glencross DK. Flow cytometric analysis of platelet surface glycoproteins in the diagnosis of Bernard-Soulier syndrome. Pediatric hematology and oncology. 1997 Jan-Feb:14(1):43-50 [PubMed PMID: 9021812]
Level 2 (mid-level) evidenceBlajchman MA, Beckers EA, Dickmeiss E, Lin L, Moore G, Muylle L. Bacterial detection of platelets: current problems and possible resolutions. Transfusion medicine reviews. 2005 Oct:19(4):259-72 [PubMed PMID: 16214015]
Gabe C, Ziza KC, Durazzo N, Pagani FM, Oliveira VB, Conrado MAV, Dezan MR, Mendrone A Jr, Villaça PR, Dinardo CL, Rocha V. Detection of alloimmunization in Glanzmann Thrombasthenia and Bernard-Soulier Syndrome: Data from a Brazilian Center. Hematology, transfusion and cell therapy. 2023 Jul:45 Suppl 2(Suppl 2):S101-S107. doi: 10.1016/j.htct.2022.06.005. Epub 2022 Jul 20 [PubMed PMID: 36114116]
Nurden AT, Freson K, Seligsohn U. Inherited platelet disorders. Haemophilia : the official journal of the World Federation of Hemophilia. 2012 Jul:18 Suppl 4():154-60. doi: 10.1111/j.1365-2516.2012.02856.x. Epub [PubMed PMID: 22726100]
Zaninetti C, Gresele P, Bertomoro A, Klersy C, De Candia E, Veneri D, Barozzi S, Fierro T, Alberelli MA, Musella V, Noris P, Fabris F, Balduini CL, Pecci A. Eltrombopag for the treatment of inherited thrombocytopenias: a phase II clinical trial. Haematologica. 2020 Mar:105(3):820-828. doi: 10.3324/haematol.2019.223966. Epub 2019 Jul 4 [PubMed PMID: 31273088]
Level 1 (high-level) evidenceEstcourt LJ, Birchall J, Allard S, Bassey SJ, Hersey P, Kerr JP, Mumford AD, Stanworth SJ, Tinegate H, British Committee for Standards in Haematology. Guidelines for the use of platelet transfusions. British journal of haematology. 2017 Feb:176(3):365-394. doi: 10.1111/bjh.14423. Epub 2016 Dec 23 [PubMed PMID: 28009056]
Ozelo MC, Svirin P, Larina L. Use of recombinant factor VIIa in the management of severe bleeding episodes in patients with Bernard-Soulier syndrome. Annals of hematology. 2005 Nov:84(12):816-22 [PubMed PMID: 16044315]
Level 3 (low-level) evidenceHacihanefioglu A, Tarkun P, Gonullu E. Use of recombinant factor VIIa in the management and prophylaxis of bleeding episodes in two patients with Bernard-Soulier syndrome. Thrombosis research. 2007:120(3):455-7 [PubMed PMID: 17141823]
Level 3 (low-level) evidenceLocatelli F, Rossi G, Balduini C. Hematopoietic stem-cell transplantation for the Bernard-Soulier syndrome. Annals of internal medicine. 2003 Jan 7:138(1):79 [PubMed PMID: 12513061]
Level 3 (low-level) evidenceRieger C, Rank A, Fiegl M, Tischer J, Schiel X, Ostermann H, Kolb HJ. Allogeneic stem cell transplantation as a new treatment option for patients with severe Bernard-Soulier Syndrome. Thrombosis and haemostasis. 2006 Jan:95(1):190-1 [PubMed PMID: 16543979]
Level 3 (low-level) evidenceReisi N. Bernard-Soulier syndrome or idiopathic thrombocytopenic purpura: A case series. Caspian journal of internal medicine. 2020 Winter:11(1):105-109. doi: 10.22088/cjim.11.1.105. Epub [PubMed PMID: 32042394]
Level 2 (mid-level) evidenceGadisseur A, Hermans C, Berneman Z, Schroyens W, Deckmyn H, Michiels JJ. Laboratory diagnosis and molecular classification of von Willebrand disease. Acta haematologica. 2009:121(2-3):71-84. doi: 10.1159/000214846. Epub 2009 Jun 8 [PubMed PMID: 19506352]
Othman M. Platelet-type von Willebrand disease and type 2B von Willebrand disease: a story of nonidentical twins when two different genetic abnormalities evolve into similar phenotypes. Seminars in thrombosis and hemostasis. 2007 Nov:33(8):780-6. doi: 10.1055/s-2007-1000368. Epub [PubMed PMID: 18175283]
Kahr WH, Dror Y. Gray platelet syndrome: macrothrombocytopenia with deficient α-granules. Blood. 2012 Sep 27:120(13):2543 [PubMed PMID: 23193541]
Level 3 (low-level) evidenceBreton-Gorius J, Favier R, Guichard J, Cherif D, Berger R, Debili N, Vainchenker W, Douay L. A new congenital dysmegakaryopoietic thrombocytopenia (Paris-Trousseau) associated with giant platelet alpha-granules and chromosome 11 deletion at 11q23. Blood. 1995 Apr 1:85(7):1805-14 [PubMed PMID: 7703487]
Shprintzen RJ. Velo-cardio-facial syndrome: 30 Years of study. Developmental disabilities research reviews. 2008:14(1):3-10. doi: 10.1002/ddrr.2. Epub [PubMed PMID: 18636631]
McDonald-McGinn DM, Sullivan KE, Marino B, Philip N, Swillen A, Vorstman JA, Zackai EH, Emanuel BS, Vermeesch JR, Morrow BE, Scambler PJ, Bassett AS. 22q11.2 deletion syndrome. Nature reviews. Disease primers. 2015 Nov 19:1():15071. doi: 10.1038/nrdp.2015.71. Epub 2015 Nov 19 [PubMed PMID: 27189754]
Nurden AT. Qualitative disorders of platelets and megakaryocytes. Journal of thrombosis and haemostasis : JTH. 2005 Aug:3(8):1773-82 [PubMed PMID: 16102044]
Level 2 (mid-level) evidenceBeales IL. An acquired-pseudo Bernard Soulier syndrome occurring with autoimmune chronic active hepatitis and anti-cardiolipin antibody. Postgraduate medical journal. 1994 Apr:70(822):305-8 [PubMed PMID: 8183781]
Level 3 (low-level) evidenceDevine DV, Currie MS, Rosse WF, Greenberg CS. Pseudo-Bernard-Soulier syndrome: thrombocytopenia caused by autoantibody to platelet glycoprotein Ib. Blood. 1987 Aug:70(2):428-31 [PubMed PMID: 3607280]
Level 3 (low-level) evidenceSehgal T, Altohami M, Lafferty N, Desborough M, Boyce S, Kazmi R, Jenner M. Acquired Bernard-Soulier syndrome and hypodysfibrinogenaemia because of multiple myeloma. Blood coagulation & fibrinolysis : an international journal in haemostasis and thrombosis. 2022 Mar 1:33(2):130-133. doi: 10.1097/MBC.0000000000001104. Epub [PubMed PMID: 34799506]
El Hammoumi M, Kabiri EH. Successful management of a retrosternal goiter in a patient with Bernard-Soulier syndrome. Kardiochirurgia i torakochirurgia polska = Polish journal of cardio-thoracic surgery. 2022 Jun:19(2):102. doi: 10.5114/kitp.2022.117500. Epub 2022 Jun 29 [PubMed PMID: 35891989]
Faheem O, Hussain B, Khurshid M, Hamid A, Rashid A. Multivessel Percutaneous Coronary Intervention in a Patient With Bernard-Soulier Syndrome. JACC. Case reports. 2020 Apr:2(4):621-625. doi: 10.1016/j.jaccas.2019.12.025. Epub 2020 Mar 25 [PubMed PMID: 34317307]
Level 3 (low-level) evidence