Introduction
Hypertrophic cardiomyopathy (HCM) is an autosomal dominant cardiac myocyte disease caused by mutations in sarcomere and sarcomere-related protein genes encoding for elements of the contractile machinery of the heart. Characteristic cardiac structural changes include increased left ventricular wall thickness (hypertrophy) causing dynamic left ventricular outflow obstruction, diastolic dysfunction, myocardial ischemia, arrhythmias, autonomic dysfunction, and mitral regurgitation. HCM is a condition that has historically been referred to as idiopathic hypertrophic subaortic stenosis. In the United States, HCM is the most common identifiable cause of sudden cardiac death in healthy people aged younger than 35, including well-trained athletes.[1]
About 60% of patients with HCM have a known gene mutation of the sarcomere or sarcomere-related genes, and obtaining a detailed family history is crucial to help risk-stratify affected patients. A comprehensive patient history and thorough physical exam can also help identify patients at risk of malignant arrhythmia. These structural and functional abnormalities can produce fatigue, dyspnea, chest pain, palpitations, and syncope.[2][3][4] Early intervention through pharmacologic means, internal cardiac defibrillator placement, and surgery, when warranted, have greatly improved the survival rates of HCM in the last few decades.
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
Familial HCM occurs as an autosomal dominant Mendelian-inherited disorder in approximately 60% of cases. Incomplete penetrance and variable expression can cause unpredictable manifestations, even within the same family. In individuals diagnosed with HCM and a known pathogenic sarcomeric gene variant, the 2 most frequently detected genes are β-myosin heavy chain 7 (MYH7) and myosin-binding protein C3 (MYBPC3), accounting for about 75% of known variant-positive patients.[5] Less common genetic variants encode thin filament proteins, such as troponin T, troponin I, myosin light chains, and actin; TNNI3, TNNT2, TPM1, MYL2, MYL3, and ACTC1 mutations make up a smaller proportion of patients, ranging from 1% to 5%. Over 1500 variants causing HCM have been identified, with most unique to each family. No clear connection has been shown between prognosis and specific gene mutation.
A significant portion of individuals diagnosed with HCM do not have a genetic explanation for their condition; this includes a subset of patients, up to 40% in some studies, who do not have any family members affected by the disease—known as “nonfamilial” HCM. These findings suggest the existence of other yet undiscovered pathophysiological mechanisms that may play a role in the manifestation of HCM pathology.[6] Recent data has also associated abnormal myocardial calcium kinetics with the cause of inappropriate myocardial hypertrophy and specific features of HCM, especially in patients with diastolic functional abnormalities.[7][8][9][10]
Epidemiology
The prevalence of HCM in the general population worldwide is 0.2% (1 in 500 adults), as determined by echocardiographic studies. Morphologic abnormalities are found in approximately 25% of first-degree relatives of patients with HCM. HCM is more common in males than females, although the condition is autosomal dominant, and genetic inheritance does not follow sex predilection. The explanation for this phenomenon may be related to screening strategies, genetics, or hormonal regulation.[11] However, there is evidence that women are diagnosed at an older age and have a worse prognosis, suggesting that diagnosis strategies should be revisited.[2][5] The most common presentation of HCM is in the third decade of life but may be present at any age, from newborns to older individuals.
Pathophysiology
Wall thickening can involve any area of the myocardium, but the interventricular septal wall is the most commonly affected location. In the absence of other conditions that could possibly contribute to left ventricular hypertrophy (LVH), such as severe hypertension or aortic stenosis, HCM is usually diagnosed by echocardiogram.
- HCM in adults is characterized by a left ventricular wall thickness of 15 mm or more, with a septal/posterior wall thickness ratio of over 1.3 in patients who are normotensive or over 1.5 in those who are hypertensive. A wall thickness of 13 to 14 mm can be diagnostic for positive family history, evidence of left ventricular outflow tract obstruction (LVOTO), or electrocardiogram (ECG) abnormalities.[12]
- In children, hypertrophy is defined as a wall thickness that is at least 2 standard deviations above the mean (z-score ≥2) for age, sex, or body size or LV wall thickness of 13 mm or more.[12]
Five mechanisms contribute to the development of HCM: dynamic LVOTO, mitral regurgitation, diastolic dysfunction, myocardial ischemia, and autonomic dysfunction.
Dynamic LVOTO: Obstruction can occur either at rest or with exercise. About one-third of patients will each have no obstruction, obstruction with exercise, or obstruction at rest. A gradient of 30 mm Hg or more is considered hemodynamically significant and is associated with death and progression to heart failure; however, a gradient of 50 mm Hg or more is regarded as a threshold for surgical and percutaneous intervention, primarily if symptomatic.[5] The gradient can also be provoked by changes in preload and afterload, as later described in the History and Physical section.
- The likelihood of obstruction is typically determined by the pattern of hypertrophy in the left ventricle, with classical asymmetric left ventricular hypertrophy (LVH) most commonly affecting the basal interventricular septum.
- LVOTO in HCM can be attributed to 2 main causes. The first is an increase in septal thickness, which narrows the LVOT and leads to abnormal movement of the mitral valve leaflets. Systolic anterior motion (SAM) of the mitral valve is common in HCM and is defined as displacement of the distal anterior mitral valve leaflet into the LVOT.[13] The second cause is alterations in the structure of the mitral valve, such as elongated leaflets and displacement of the papillary muscles and valve apparatus towards the anterior region. These structural changes render the valve liable to unnatural flow vectors. SAM is more common with a hyperdynamic left ventricle; the ejection fraction is often elevated in HCM (>65%).[14]
Mitral Regurgitation: This can be due to LVOTO or SAM of the mitral valve. The regurgitant jet direction can guide the causative mechanism, and SAM is usually directed posteriorly.[5]
Diastolic Dysfunction: The presence of LVH in conjunction with myocardial disarray and fibrosis increases left ventricular stiffness. This, in turn, negatively affects myocardial relaxation and diastolic function, ultimately leading to increased intracavitary pressures in both the left ventricle (LV) and left atrium (LA). Delayed inactivation from abnormal intracellular calcium reuptake has also been observed in patients with HCM.
Myocardial Ischemia: Common findings in HCM include myocardial hypertrophy, microvascular dysfunction, impaired coronary flow reserve, and medial and intimal hypertrophy of intramural arterioles. All these factors contribute to a mismatch between myocardial oxygen supply and demand, resulting in myocardial fibrosis.
Autonomic Dysfunction: Autonomic dysfunction in HCM is defined as an abnormal blood pressure response to exercise—specifically, a failure to increase systolic blood pressure by at least 20 mm Hg or a drop in systolic blood pressure during exercise of more than 20 mm Hg from the peak value—is associated with poor prognosis. The proposed mechanism involves the inappropriate firing of stretch-sensitive mechanoreceptors in the LV myocardium, resulting in unreciprocated changes in systemic vascular resistance, which may lead to episodes of presyncope or syncope.
The most common arrhythmia in HCM is atrial fibrillation, present in about 25% of patients, which is 4- to 6-fold higher than in the general population. Atrial fibrillation is poorly tolerated in patients with HCM. Due to decreased LV compliance, up to 35% of left ventricular filling depends on left atrial contraction. The decreased LV filling can lead to lower cardiac output and hypotension.[7] Non-sustained ventricular tachycardia (NSVT) is present in 20% to 30% of patients with HCM. NSVT can lead to ventricular fibrillation—the most common cause of sudden cardiac death (SCD). Potential etiology of the arrhythmias includes interstitial fibrosis, myocyte disarray, and myocardial ischemia.[14]
Histopathology
The histology of HCM provides crucial insights into the structural changes occurring within the myocardium. HCM is characterized by enlarged cardiomyocytes in the heart muscle, which have eosinophilic cytoplasm and box-shaped nuclei. These cells can exhibit unusual shapes, such as Y-shaped branches, frequent side-to-side connections, or a spiraled appearance around a central fibrous core. The resulting disorganized architecture of the heart muscle can lead to myocardial disarray, with bundles of cardiomyocytes arranged at different angles to each other.
The affected areas can be adjacent to areas of normal myocardium in a patchy nature. The disarray is inversely correlated to fibrosis. Fibrosis is seen interstitially and will stain blue with the Masson trichrome stain.[5][14] Microvascular ischemia can cause cell death and replacement fibrosis, while interstitial fibrosis can lead to arrhythmogenic foci. HCM is also associated with intercellular junction abnormalities in the intercalated discs and desmosomes, which can form “huge mega-discs” that likely contribute to diastolic dysfunction and arrhythmias.[5]
History and Physical
Up to 50% of individuals diagnosed with HCM may not experience any symptoms or may only have mild symptoms—especially patients without LVOTO.[12] The patient history should focus on both family and personal history:
- Family history of HCM, sudden cardiac death, unexplained syncope
- Personal history of chest pain, syncope, unexplained dizziness, palpitations, shortness of breath, chest pain
Shortness of breath and chest discomfort are often caused by underlying diastolic dysfunction and LVOTO. Chest pain can present as typical or atypical angina. Individuals with LVOTO may experience more severe symptoms after consuming a heavy meal (postprandial exacerbation). Dizziness and syncope can occur at rest or during physical exertion, and arrhythmia or dynamic LVOTO can be the underlying causes. Palpitations are also common and may indicate underlying episodes of atrial fibrillation or ventricular arrhythmia. Sudden cardiac death represents the most devastating presenting symptom.
Physical examination findings are characterized by a “jerky” pulse with rapid upstroke and downstroke, a normal S1 and split S2, an S3 due to decompensated heart failure, prominent “a wave” of jugular venous pressure, laterally displaced apical impulse, and double carotid pulse. An S4 is also common with LVH.
A harsh mid-systolic ejection murmur at the left sternal border is affected as follows:
- Diminished intensity with increased preload (squatting) or afterload (handgrip)
- Increased intensity with a decrease in preload (Valsalva maneuver, standing) or with any reduction in afterload (vasodilator administration)
Exercise will also increase the murmur. If mitral regurgitation is present, a holosystolic, blowing murmur may be loudest at the apex.
Evaluation
Individuals with a family history of HCM, cardiac symptoms or a cardiac event, a heart murmur revealed during a routine physical examination, or an abnormal 12-lead ECG may require further clinical assessment. To accurately evaluate for HCM, a comprehensive cardiac history, including family history spanning 3 generations, and an extensive physical examination (incorporating special provocative maneuvers such as Valsalva, squat-to-stand, or passive leg raising) are necessary. An ECG and cardiac imaging to identify LVH should be performed in all patients.[15][16][17]
Laboratory testing: Lab testing, including Troponin I and T, can be used to detect suspected ischemic events. Elevation of the natriuretic peptides, especially those of the brain, can also be used as a marker of heart failure. Elevated NT-pro-BNP levels are correlated with LV wall stress and diastolic and systolic dysfunction.
Electrocardiogram: ECG is often abnormal, even if LVOTO is not present. Findings include localized or widespread repolarization changes, prominent Q-waves in the inferior (II, III, and aVF) and lateral (I, aVL, and V4-V6) leads, left atrial or biatrial enlargement, left axis deviation, and deeply inverted T-waves. Prominent Q-waves can represent thickened interventricular septal depolarization.[14]
Transthoracic echocardiogram: This echocardiogram can demonstrate the typical cardiac morphology of unexplained LVH with a wall thickness of more than 15 mm. About one-third of patients with HCM will also have increased RV wall thickness (>8 mm). As noted above, a septal/posterior wall thickness ratio over 1.3 in normotensive patients or over 1.5 in hypertensive patients is also diagnostic of HCM. Transthoracic echocardiogram (TTE) can also show systolic and diastolic function, the presence and severity of any LVOT gradient, and the degree of mitral regurgitation or SAM (of note, SAM has a specificity of 99% for HCM). Left atrial measurements and pulmonary artery pressures should also be imaged. TTE with contrast can also be performed in patients unable to have a cardiac magnetic resonance imaging scan, such as if an implantable cardioverter-defibrillator (ICD) is present (see Image. Hypertrophic Cardiomyopathy, Echocardiogram).
Ambulatory ECG monitoring (Holter): Monitoring should be performed for 24 to 48 hours in all patients diagnosed with HCM for risk assessment of ventricular arrhythmias and sudden death.
Exercise stress testing: This can be performed for risk stratification and LVOT gradient assessment. Exercise stress testing with TTE is the preferred method to image the LVOT because this simulates physiologic stress.[18]
Cardiac magnetic resonance: The cardiac magnetic resonance (CMR) is becoming more commonly used to characterize cardiac tissue and structure in patients with HCM and their relatives. With gadolinium, CMR can also detect myocardial fibrosis, an important prognostic indicator. Late gadolinium enhancement (LGE) is present in over 60% of patients with symptomatic HCM and is thought to be a marker for replacement fibrosis.[5]
- The American Heart Association’s (AHA) guidelines suggest using CMR in the following situations:
- For patients in whom TTE is inconclusive, or images are suboptimal
- For patients diagnosed with LVH who may have alternative diagnoses, such as Fabry disease, amyloidosis, or athlete’s heart
- For patients with unclear benefits regarding ICD placement
- TTE can miss patients with the uncommon apical or anterolateral wall hypertrophy forms of HCM, so CMR is preferred for imaging this variant.
- To image more subtle structural findings such as myocardial crypts, elongated mitral leaflets, and expanded extracellular space [12]
- To define the LVOT when planning invasive intraseptal reduction treatments [19][20]
Some patients may also require additional studies as follows:
- Cardiac catheterization: This catheterization can help characterize cardiac hemodynamics, the degree of left ventricular outflow obstruction, and the coronary vessel anatomy.
- Electrophysiological studies: These studies can help determine the origin of the arrhythmias.
Risk Stratification in Young Athletes and Patients
HCM is a leading cause of sudden cardiac death (SCD) in athletes younger than 35 years (along with congenital anomalies of the coronary arteries and arrhythmogenic right ventricular cardiomyopathy [ARVC]). For most patients with HCM, engaging in mild-to-moderate-intensity recreational exercise can be beneficial. Special attention is paid to athletes because about 25% of SCD cases occur during physical activity, such as sports.[21] Risk stratification for young athletes to evaluate for HCM is performed using ECG and echocardiography. However, these testing modalities have low sensitivity, so CMR has been suggested as a test for all young athletes with a personal history of syncope, a family history of syncope or SCD, or an abnormal or indeterminate echocardiogram.[8] For athletes older than 35 years, coronary artery disease is the most common cause of SCD.
In the United States, HCM and an anomalous origin of coronary arteries are the most common causes of SCD in all patients younger than 35, while in Europe, arrhythmogenic cardiomyopathies (including ARVC) and channelopathies are more common.[22][23] Current guidelines recommend against competitive sports and strenuous exercise, given the association of SCD in young athletes with HCM.[12] However, the prospective, observational LIVE-HCM trial studying the effect of vigorous exercise did not find an association with life-threatening arrhythmia or death. Results from this study found that most significant cardiac events occurred during activities of daily living.[24] Another ongoing trial at Yale University (NCT02549664) is also studying the effect of exercise on patients with HCM. As no well-designed trials have definitively proven that avoiding strenuous exercise helps decrease the chances of SCD, some experts recommend shared decision-making between patients and physicians after fully explaining all possible risks and benefits.[21][22]
Treatment / Management
Treatment strategies for HCM are based on observational data and clinical experience since no large randomized trials have been performed. Pharmacological therapy is the first-line approach to symptomatic HCM. Initial recommended medications include negative inotropic agents, including beta blockers, nondihydropyridine calcium channel blockers, and disopyramide.[25][26](B3)
Medical Intervention
- Mavacamten is a first-in-class drug approved in 2022 by the Food and Drug Administration that is specifically for the treatment of obstructive HCM. This medicine is the first selective, allosteric, reversible small-molecule adenosine triphosphatase inhibitor of cardiac myosin that functions by blocking the interaction between myosin and actin within cardiac muscle cells, which exerts a negative inotropic effect. This mechanism helps to relieve the obstruction in the left ventricular outflow tract, thereby enhancing blood flow and alleviating symptoms. Mavacamten is presently indicated for the treatment of adults with symptomatic heart failure (New York Heart Association [NYHA] class II and III) due to obstructive HCM. Typical side effects include dizziness, fainting, and shortness of breath. Severe side effects may involve heart failure that requires close monitoring and possible dosage adjustment if the left ventricular ejection fraction drops below 50%.
- Beta-blockers are the first line of management for most HCM cases. Patients should be titrated to a therapeutic dose that provides symptom relief. Beta-blockade failure should not be declared until suppression of the resting heart rate is demonstrated. This approach ensures that patients receive optimal treatment benefits while minimizing the risk of adverse effects.
- Calcium channel blockers, such as diltiazem and verapamil, are known to alleviate symptoms in patients with obstructive HCM. These drugs can have vasodilating properties, but they can also have negative inotropic and negative chronotropic effects, which may limit their use. Evidence does not support using calcium channel blockers in combination with beta-blockers as a therapy for HCM.
- Disopyramide is a sodium channel blocker classified as a class Ib antiarrhythmic drug; this is also a potent negative inotropic agent that is used off-label for the treatment of HCM. Disopyramide helps reduce outflow obstruction and associated symptoms. As cytochrome P450 3A4 metabolizes this drug, many drugs, such as calcium channel blockers, can interact with disopyramide and affect its serum levels. Due to the anticholinergic properties of disopyramide, it can potentially cause urinary retention and exacerbate related conditions like glaucoma and myasthenia gravis.
- Diuretics can be given carefully in patients without LVOT obstruction who have refractory heart failure symptoms presenting with volume overload. Volume depletion decreases stroke volume and worsens the LVOT gradient. This can lead to hypotension, lightheadedness, and syncope.
- Certain medications should be avoided or used very cautiously as they can worsen symptoms. These include vasodilators, amlodipine, nifedipine, nitroglycerin, angiotensin-converting enzyme/angiotensin II receptor blockers, and adrenergic receptor β-agonists like dopamine and dobutamine.[27]
- Patients with nonobstructed HCM are usually treated medically.[18][28] (A1)
Implantable Cardiac-Defibrillator Placement
- The placement of an implantable cardiac-defibrillator (ICD) is considered largely responsible for the improved mortality of HCM over the last 20 years.
- Any patient with risk factors for sudden cardiac death should be considered for ICD. Risk factors include the following: recent unexplained syncope, HCM-related SCD in a close relative, thin-walled akinetic or dyskinetic LV apical aneurysm with scarring, frequent or prolonged episodes of ventricular tachycardia on ambulatory monitoring, extensive late gadolinium enhancement on CMR, and massive LVH (>30 mm).
- Multiple study results have shown primary ICD placement terminates 3% to 4% of malignant tachyarrhythmias per year, and that number rises to 10% for secondary ICD placement (after cardiac arrest).[27]
- Although overall beneficial, the complication rate can be up to 10% per year, including lead fractures and infections.[27]
- Guidelines for ICD placement in children are less clear as children have less marginal benefit and increased complications from ICDs than adults due to smaller size.[21]
Surgical intervention
Up to 50% of patients with HCM may be refractory to medical treatment.[29] Indications for surgical intervention include New York Heart Association III/IV class symptoms despite optimal medical therapy, syncope related to hemodynamic compromise from LVOT obstruction, or an LVOT gradient of more than 50 mm Hg.
- Septal reduction therapy includes left ventricular septal myectomy, which immediately reduces the outflow tract obstruction. Mitral valve repair or remodeling is sometimes also performed to reduce mitral regurgitation or further relieve LVOTO. Complete heart block requiring a permanent pacemaker is a myectomy complication occurring 1% to 5% of the time. Mortality is low with experienced surgeons.[27]
- Alcohol septal ablation can also be performed for the same indications and has the added benefit of faster recovery time. However, the LVOT obstruction often takes up to 3 months to show improvement, and results are less uniform than with surgical myectomy. In addition, the risk of complete heart block is higher (10% to 15%), and a post-ablation scar can form, causing an arrhythmogenic focus.[30]
- Given that atrial fibrillation is the most common arrhythmia associated with HCM, a Maze procedure is often done in conjunction if a myectomy is required, and it is often successful in decreasing or resolving this arrhythmia.
- Radiofrequency ablation is a newer technique being studied for left ventricular septal reduction. Results are similarly effective to the more established surgical myectomy and alcohol ablation techniques. The most common adverse effect is pericardial effusion requiring drainage. Further studies of this technique are needed.[29][31]
- A heart transplant is recommended as a last resort in patients who have failed all medical and surgical treatments (see Image. Hypertrophic Cardiomyopathy Treatment Mapping).[27][32]
Differential Diagnosis
For a correct diagnosis of HCM to be made, other diagnoses must be excluded first, including the following:
Fabry disease: This disease is transmitted through X-linked inheritance and is caused by an α-galactosidase deficiency characterized by angiokeratomas and peripheral neuropathies.
LEOPARD syndrome: Patients with this syndrome exhibit multiple lentigines.
Amyloidosis: Patients with ventricular hypertrophy due to amyloidosis will most likely have bilateral carpal tunnel syndrome, nephrotic syndrome, or skin abnormalities. The ECG may show an arteriovenous block, and the echocardiogram may show a “sparkling” appearance.
Danon disease: This is an X-linked lysosomal glycogen-storage disease with the clinical triad of skeletal myopathy, cardiomyopathy, and intellectual disability.
Friedrich ataxia: This is the most common cause of inherited ataxia caused by a mutation in the frataxin gene, a mitochondrial protein found in neurons and cardiomyocytes. Ataxia, dysarthria, and other cerebellar signs usually become prominent during childhood.
Aortic stenosis or insufficiency/pulmonary hypertension: These clinical entities combine LVH with aortic valve or pulmonary artery changes and can cause symmetric or asymmetric LVH, similar to HCM. Abnormalities may be detectable on echocardiogram.
Athlete’s heart: A history of athletic achievement and strenuous training should help in differential diagnosis. An echocardiogram may show a low-normal ejection fraction, and all the heart chambers may be enlarged instead of just the LV. Contractility reserve is increased, but this may not be apparent on echocardiography. CMR is a valuable tool to differentiate this from HCM, as athlete’s heart will not show LGE (late gadolinium enhancement).[33][34]
Hypertensive heart disease: Diffuse atherosclerotic changes and a history of essential hypertension should suggest the diagnosis. HCM is also associated with systolic anterior motion, late gadolinium enhancement, and preferential involvement of the apical, anterolateral free wall or posterior septum, which is usually not seen in hypertensive heart disease.
Prognosis
Understanding the prognosis of HCM is essential for guiding patient care and counseling. Factors such as age of onset, genetic mutations, degree of hypertrophy, presence of symptoms, and complications significantly influence the long-term outlook. A nuanced understanding of these prognostic indicators is crucial for clinicians to tailor treatment strategies and optimize patient outcomes.
- Previously, mortality rates of 1% to 4% were reported in patients with HCM. With appropriate treatment, these numbers are now thought to be closer to 0.5%.
- Even though many patients with HCM are asymptomatic, the first clinical presentation may be sudden death from malignant arrhythmias.
- Increased left atrial size (transverse linear dimension >48 mm or volume >118 mL) is correlated with an increased risk of death, heart failure, and atrial fibrillation.[5]
Complications
Complications of HCM pose significant challenges in case management and prognostication. Understanding the diverse array of complications associated with HCM is essential for clinicians to implement appropriate preventive measures and therapeutic interventions, ultimately improving patient outcomes and quality of life.
These complications encompass a spectrum of cardiovascular events, including the following:
- Ventricular arrhythmias
- Congestive heart failure
- Infective endocarditis of the mitral valve
- Atrial fibrillation
- Embolic phenomenon
- Sudden death
Deterrence and Patient Education
Preventing and deterring the progression of HCM is paramount in reducing its associated morbidity and mortality. Lifestyle modifications and targeted interventions can be crucial in mitigating its impact. Understanding the strategies for deterrence and prevention is vital for healthcare professionals to empower patients with HCM and optimize their long-term cardiac health. Patients should be screened and advised on preventing and treating comorbidities that can exacerbate the severity of HCM. These comorbidities include atherosclerotic cardiovascular disease, obesity, hypertension, and sleep apnea.
Individuals with a history of sudden death in a family member within 3 generations should be screened for the disorder. Unless symptomatic, screening by ECG, echocardiography, or CMR usually begins at 12 years and continues yearly until age 21, after which screening can occur every 3 to 5 years. Genetic testing can be performed if a gene causing HCM can be identified in a proband, but this is not the preferred strategy per the Journal of the American College of Cardiology due to variable genetic penetrance and expression, as well as the absence of identifiable genetic mutations in up to 40% of HCM cases.[12] Individuals with abnormal blood pressure in response to exercise should also be screened. Significant controversy exists about the safety of participating in organized or competitive sports in patients with HCM. Associated risks, particularly sudden cardiac death, should be discussed in depth with affected patients and caregivers.
Pearls and Other Issues
Additional pertinent information includes:
- Early diagnosis of HCM is important as it allows the healthcare professional to prescribe appropriate treatment.
- If a patient is diagnosed with HCM, close family members should be screened.
- Mutations in β-myosin heavy chain 7 (MYH7) and myosin-binding protein C3 (MYBPC3) account for about 75% of known variant-positive patients.
- Systolic anterior motion is highly suggestive of HCM and is associated with mitral regurgitation.
- Those who have concomitant mitral regulation and diastolic dysfunction are also prone to recurrent episodes of heart failure.
- Atrial fibrillation is the most common arrhythmia associated with HCM.
- Ventricular tachycardia degenerating into ventricular fibrillation is the most common cause of SCD.
- The most common identifiable cause of SCD in the United States is HCM. In Europe, the most common cause is arrhythmogenic right ventricular cardiomyopathy.
- Screening and monitoring are usually performed through a combination of ECG, echocardiogram, and CMR.
- The increased use of ICDs is thought to be the reason for improved survival in patients with HCM.
- Surgical myectomy and alcohol ablation are 2 well-established techniques for reducing the left ventricular septal wall thickness and decreasing LVOTO.
Enhancing Healthcare Team Outcomes
Patients with HCM are at high risk of SCD; therefore, early identification and management of HCM cases are imperative in reducing morbidity and mortality. The care of patients with HCM necessitates a collaborative approach among healthcare professionals to ensure patient-centered care and improve overall outcomes. Cardiologists, electrophysiologists, primary care physicians, emergency medicine physicians, radiologists, geneticists, advanced practitioners, nurses, pharmacists, and other healthcare professionals involved in the care of these patients should possess the essential clinical skills and knowledge to diagnose and manage HCM accurately. This includes expertise in recognizing the varied clinical presentations and understanding the nuances of diagnostic techniques such as ECG, echocardiogram, and CMR. Patient and caregiver education about disease progression, medication compliance, possible risks and benefits of exercise, and the importance of cardiac follow-up is crucial for optimal outcomes.
A strategic approach involving evidence-based strategies to optimize treatment plans and minimize adverse effects is equally crucial. Ethical considerations must guide decision-making, ensuring informed consent and respecting patient autonomy in treatment choices, especially given the association with SCD and the unpredictable nature of this disease. Each healthcare professional must be aware of their responsibilities and contribute their unique expertise to the patient’s care plan, fostering a multidisciplinary approach. Effective interprofessional communication is paramount, allowing seamless information exchange and collaborative decision-making among the team members. Care coordination plays a pivotal role in ensuring that the patient’s journey from diagnosis to treatment and follow-up is well-managed, minimizing errors and enhancing patient safety. By embracing these principles of skill, strategy, ethics, responsibilities, interprofessional communication, and care coordination, the healthcare team can deliver patient-centered care, ultimately improving patient outcomes and enhancing team performance.
Media
(Click Image to Enlarge)
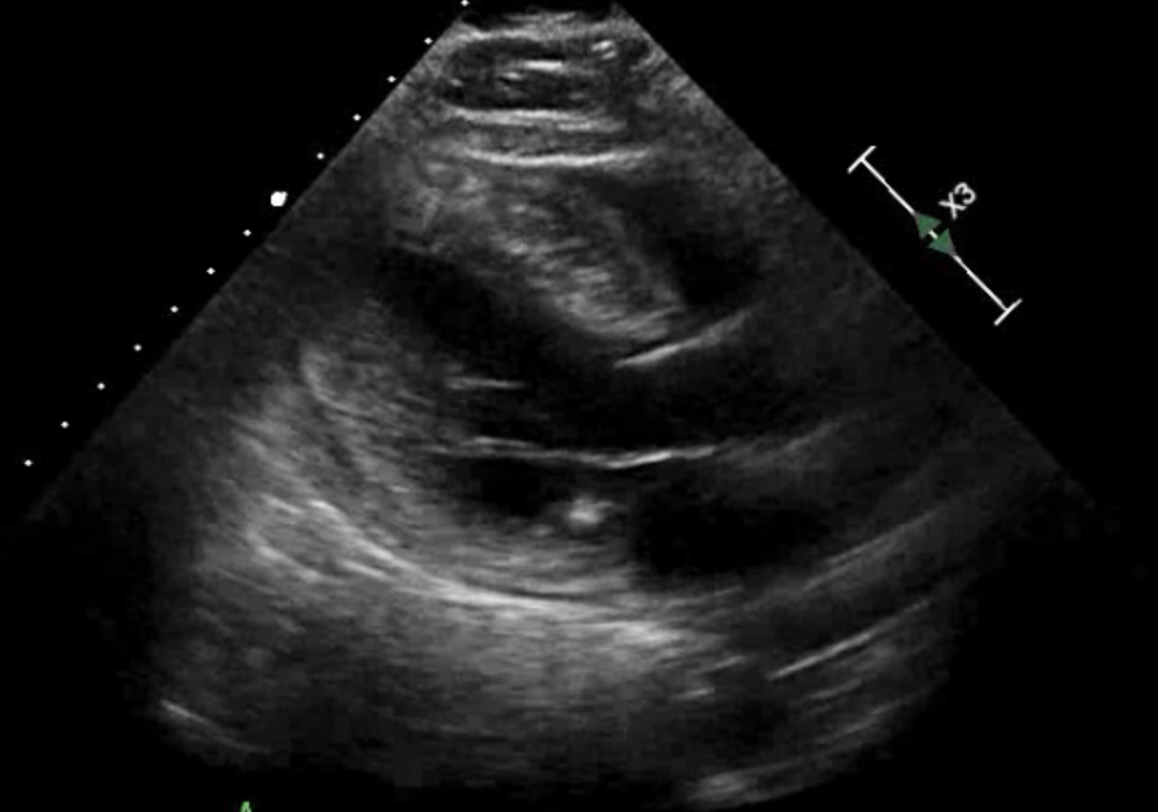
Hypertrophic Cardiomyopathy, Echocardiogram. The image illustrates hypertrophic cardiomyopathy, a condition affecting the left ventricle of the heart.
Contributed by Y Hafeez, DO
(Click Image to Enlarge)

Hypertrophic Cardiomyopathy Treatment Mapping. The diagram illustrates treatment mapping adopted from the American Heart Association guidelines, 2020. HCM, hypertrophic cardiomyopathy; NSVT, non-sustained ventricular tachycardia; CRT, cardiac resynchronization therapy, NYHA, New York Heart Association; GDMT, guideline-directed medical therapy; LBBB, left bundle branch block; ICD, Implantable cardioverter defibrillator
Contributed by MH Alahmadi, MBBS, MS
References
Czimbalmos C, Csecs I, Toth A, Kiss O, Suhai FI, Sydo N, Dohy Z, Apor A, Merkely B, Vago H. The demanding grey zone: Sport indices by cardiac magnetic resonance imaging differentiate hypertrophic cardiomyopathy from athlete's heart. PloS one. 2019:14(2):e0211624. doi: 10.1371/journal.pone.0211624. Epub 2019 Feb 14 [PubMed PMID: 30763323]
Spudich JA. Three perspectives on the molecular basis of hypercontractility caused by hypertrophic cardiomyopathy mutations. Pflugers Archiv : European journal of physiology. 2019 May:471(5):701-717. doi: 10.1007/s00424-019-02259-2. Epub 2019 Feb 15 [PubMed PMID: 30767072]
Level 3 (low-level) evidencevan Driel B, Nijenkamp L, Huurman R, Michels M, van der Velden J. Sex differences in hypertrophic cardiomyopathy: new insights. Current opinion in cardiology. 2019 May:34(3):254-259. doi: 10.1097/HCO.0000000000000612. Epub [PubMed PMID: 30747730]
Level 3 (low-level) evidenceKraft T, Montag J. Altered force generation and cell-to-cell contractile imbalance in hypertrophic cardiomyopathy. Pflugers Archiv : European journal of physiology. 2019 May:471(5):719-733. doi: 10.1007/s00424-019-02260-9. Epub 2019 Feb 11 [PubMed PMID: 30740621]
Popa-Fotea NM, Micheu MM, Bataila V, Scafa-Udriste A, Dorobantu L, Scarlatescu AI, Zamfir D, Stoian M, Onciul S, Dorobantu M. Exploring the Continuum of Hypertrophic Cardiomyopathy-From DNA to Clinical Expression. Medicina (Kaunas, Lithuania). 2019 Jun 23:55(6):. doi: 10.3390/medicina55060299. Epub 2019 Jun 23 [PubMed PMID: 31234582]
Wijnker PJM, Sequeira V, Kuster DWD, Velden JV. Hypertrophic Cardiomyopathy: A Vicious Cycle Triggered by Sarcomere Mutations and Secondary Disease Hits. Antioxidants & redox signaling. 2019 Aug 1:31(4):318-358. doi: 10.1089/ars.2017.7236. Epub 2018 Apr 11 [PubMed PMID: 29490477]
Philipson DJ, Rader F, Siegel RJ. Risk factors for atrial fibrillation in hypertrophic cardiomyopathy. European journal of preventive cardiology. 2021 May 22:28(6):658-665. doi: 10.1177/2047487319828474. Epub [PubMed PMID: 30727760]
Mavrogeni SI, Tsarouhas K, Spandidos DA, Kanaka-Gantenbein C, Bacopoulou F. Sudden cardiac death in football players: Towards a new pre-participation algorithm. Experimental and therapeutic medicine. 2019 Feb:17(2):1143-1148. doi: 10.3892/etm.2018.7041. Epub 2018 Nov 30 [PubMed PMID: 30679986]
Maron MS, Wells S. Myocardial Strain in Hypertrophic Cardiomyopathy: A Force Worth Pursuing? JACC. Cardiovascular imaging. 2019 Oct:12(10):1943-1945. doi: 10.1016/j.jcmg.2018.09.026. Epub 2019 Jan 16 [PubMed PMID: 30660525]
Rigopoulos AG, Ali M, Abate E, Matiakis M, Melnyk H, Mavrogeni S, Leftheriotis D, Bigalke B, Noutsias M. Review on sudden death risk reduction after septal reduction therapies in hypertrophic obstructive cardiomyopathy. Heart failure reviews. 2019 May:24(3):359-366. doi: 10.1007/s10741-018-09767-w. Epub [PubMed PMID: 30617667]
Authors/Task Force members, Elliott PM, Anastasakis A, Borger MA, Borggrefe M, Cecchi F, Charron P, Hagege AA, Lafont A, Limongelli G, Mahrholdt H, McKenna WJ, Mogensen J, Nihoyannopoulos P, Nistri S, Pieper PG, Pieske B, Rapezzi C, Rutten FH, Tillmanns C, Watkins H. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: the Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). European heart journal. 2014 Oct 14:35(39):2733-79. doi: 10.1093/eurheartj/ehu284. Epub 2014 Aug 29 [PubMed PMID: 25173338]
Maron BJ, Desai MY, Nishimura RA, Spirito P, Rakowski H, Towbin JA, Rowin EJ, Maron MS, Sherrid MV. Diagnosis and Evaluation of Hypertrophic Cardiomyopathy: JACC State-of-the-Art Review. Journal of the American College of Cardiology. 2022 Feb 1:79(4):372-389. doi: 10.1016/j.jacc.2021.12.002. Epub [PubMed PMID: 35086660]
Lasala JD, Tsai J, Rodriguez-Restrepo A, Atay SM, Sepesi B. Systolic anterior motion of the mitral valve-the mechanism of postural hypotension following left intrapericardial pneumonectomy. Journal of thoracic disease. 2017 Apr:9(4):E354-E357. doi: 10.21037/jtd.2017.03.117. Epub [PubMed PMID: 28523177]
Marian AJ, Braunwald E. Hypertrophic Cardiomyopathy: Genetics, Pathogenesis, Clinical Manifestations, Diagnosis, and Therapy. Circulation research. 2017 Sep 15:121(7):749-770. doi: 10.1161/CIRCRESAHA.117.311059. Epub [PubMed PMID: 28912181]
Walsh R, Mazzarotto F, Whiffin N, Buchan R, Midwinter W, Wilk A, Li N, Felkin L, Ingold N, Govind R, Ahmad M, Mazaika E, Allouba M, Zhang X, de Marvao A, Day SM, Ashley E, Colan SD, Michels M, Pereira AC, Jacoby D, Ho CY, Thomson KL, Watkins H, Barton PJR, Olivotto I, Cook SA, Ware JS. Quantitative approaches to variant classification increase the yield and precision of genetic testing in Mendelian diseases: the case of hypertrophic cardiomyopathy. Genome medicine. 2019 Jan 29:11(1):5. doi: 10.1186/s13073-019-0616-z. Epub 2019 Jan 29 [PubMed PMID: 30696458]
Level 3 (low-level) evidenceAfanasyev A, Bogachev-Prokophiev A, Lenko E, Sharifulin R, Ovcharov M, Kozmin D, Karaskov A. Myectomy with mitral valve repair versus replacement in adult patients with hypertrophic obstructive cardiomyopathy: a systematic review and meta-analysis. Interactive cardiovascular and thoracic surgery. 2019 Mar 1:28(3):465-472. doi: 10.1093/icvts/ivy269. Epub [PubMed PMID: 30184144]
Level 1 (high-level) evidenceRobyns T, Nuyens D, Lu HR, Gallacher DJ, Vandenberk B, Garweg C, Ector J, Pagourelias E, Van Cleemput J, Janssens S, Willems R. Prognostic value of electrocardiographic time intervals and QT rate dependence in hypertrophic cardiomyopathy. Journal of electrocardiology. 2018 Nov-Dec:51(6):1077-1083. doi: 10.1016/j.jelectrocard.2018.09.005. Epub 2018 Sep 12 [PubMed PMID: 30497734]
Ommen SR, Mital S, Burke MA, Day SM, Deswal A, Elliott P, Evanovich LL, Hung J, Joglar JA, Kantor P, Kimmelstiel C, Kittleson M, Link MS, Maron MS, Martinez MW, Miyake CY, Schaff HV, Semsarian C, Sorajja P. 2020 AHA/ACC Guideline for the Diagnosis and Treatment of Patients With Hypertrophic Cardiomyopathy: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation. 2020 Dec 22:142(25):e558-e631. doi: 10.1161/CIR.0000000000000937. Epub 2020 Nov 20 [PubMed PMID: 33215931]
Level 1 (high-level) evidenceWang J, Yang S, Ma X, Zhao K, Yang K, Yu S, Yin G, Dong Z, Song Y, Cui C, Li J, Wang C, Hao J, Lu M, Chen X, Zhao S. Assessment of late gadolinium enhancement in hypertrophic cardiomyopathy improves risk stratification based on current guidelines. European heart journal. 2023 Dec 1:44(45):4781-4792. doi: 10.1093/eurheartj/ehad581. Epub [PubMed PMID: 37795986]
Lee HJ, Kim HK, Lee SC, Kim J, Park JB, Lee SP, Kim YJ. Performance of 2020 AHA/ACC HCM Guidelines and Incremental Value of Myocardial Strain for Predicting SCD. JACC. Asia. 2024 Jan:4(1):10-22. doi: 10.1016/j.jacasi.2023.09.002. Epub 2023 Oct 31 [PubMed PMID: 38222259]
Markwerth P, Bajanowski T, Tzimas I, Dettmeyer R. Sudden cardiac death-update. International journal of legal medicine. 2021 Mar:135(2):483-495. doi: 10.1007/s00414-020-02481-z. Epub 2020 Dec 21 [PubMed PMID: 33349905]
D'Ascenzi F, Valentini F, Pistoresi S, Frascaro F, Piu P, Cavigli L, Valente S, Focardi M, Cameli M, Bonifazi M, Metra M, Mondillo S. Causes of sudden cardiac death in young athletes and non-athletes: systematic review and meta-analysis: Sudden cardiac death in the young. Trends in cardiovascular medicine. 2022 Jul:32(5):299-308. doi: 10.1016/j.tcm.2021.06.001. Epub 2021 Jun 22 [PubMed PMID: 34166791]
Level 1 (high-level) evidenceAckerman M, Atkins DL, Triedman JK. Sudden Cardiac Death in the Young. Circulation. 2016 Mar 8:133(10):1006-26. doi: 10.1161/CIRCULATIONAHA.115.020254. Epub [PubMed PMID: 26951821]
Lampert R, Ackerman MJ, Marino BS, Burg M, Ainsworth B, Salberg L, Tome Esteban MT, Ho CY, Abraham R, Balaji S, Barth C, Berul CI, Bos M, Cannom D, Choudhury L, Concannon M, Cooper R, Czosek RJ, Dubin AM, Dziura J, Eidem B, Emery MS, Estes NAM, Etheridge SP, Geske JB, Gray B, Hall K, Harmon KG, James CA, Lal AK, Law IH, Li F, Link MS, McKenna WJ, Molossi S, Olshansky B, Ommen SR, Saarel EV, Saberi S, Simone L, Tomaselli G, Ware JS, Zipes DP, Day SM, LIVE Consortium. Vigorous Exercise in Patients With Hypertrophic Cardiomyopathy. JAMA cardiology. 2023 Jun 1:8(6):595-605. doi: 10.1001/jamacardio.2023.1042. Epub [PubMed PMID: 37195701]
Marrakchi S, Kammoun I, Bennour E, Laroussi L, Kachboura S. Risk stratification in hypertrophic cardiomyopathy. Herz. 2020 Feb:45(1):50-64. doi: 10.1007/s00059-018-4700-8. Epub 2018 Apr 25 [PubMed PMID: 29696341]
Daubert C, Gadler F, Mabo P, Linde C. Pacing for hypertrophic obstructive cardiomyopathy: an update and future directions. Europace : European pacing, arrhythmias, and cardiac electrophysiology : journal of the working groups on cardiac pacing, arrhythmias, and cardiac cellular electrophysiology of the European Society of Cardiology. 2018 Jun 1:20(6):908-920. doi: 10.1093/europace/eux131. Epub [PubMed PMID: 29106577]
Level 3 (low-level) evidenceMaron BJ, Desai MY, Nishimura RA, Spirito P, Rakowski H, Towbin JA, Dearani JA, Rowin EJ, Maron MS, Sherrid MV. Management of Hypertrophic Cardiomyopathy: JACC State-of-the-Art Review. Journal of the American College of Cardiology. 2022 Feb 1:79(4):390-414. doi: 10.1016/j.jacc.2021.11.021. Epub [PubMed PMID: 35086661]
Ommen SR, Mital S, Burke MA, Day SM, Deswal A, Elliott P, Evanovich LL, Hung J, Joglar JA, Kantor P, Kimmelstiel C, Kittleson M, Link MS, Maron MS, Martinez MW, Miyake CY, Schaff HV, Semsarian C, Sorajja P. 2020 AHA/ACC Guideline for the Diagnosis and Treatment of Patients With Hypertrophic Cardiomyopathy: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Journal of the American College of Cardiology. 2020 Dec 22:76(25):e159-e240. doi: 10.1016/j.jacc.2020.08.045. Epub 2020 Nov 20 [PubMed PMID: 33229116]
Level 1 (high-level) evidenceZhou M, Ta S, Hahn RT, Hsi DH, Leon MB, Hu R, Zhang J, Zuo L, Li J, Wang J, Wang B, Zhu X, Liu J, Han Y, Li X, Xu B, Zhang L, Hou L, Han C, Liu J, Liu L. Percutaneous Intramyocardial Septal Radiofrequency Ablation in Patients With Drug-Refractory Hypertrophic Obstructive Cardiomyopathy. JAMA cardiology. 2022 May 1:7(5):529-538. doi: 10.1001/jamacardio.2022.0259. Epub [PubMed PMID: 35353129]
Batzner A, Pfeiffer B, Neugebauer A, Aicha D, Blank C, Seggewiss H. Survival After Alcohol Septal Ablation in Patients With Hypertrophic Obstructive Cardiomyopathy. Journal of the American College of Cardiology. 2018 Dec 18:72(24):3087-3094. doi: 10.1016/j.jacc.2018.09.064. Epub [PubMed PMID: 30545446]
He YG, Dong Y, Yang SH, Yang F, Yin JL, Zhao HQ, Zhao YJ. Short time effects of two radiofrequency ablation methods on hypertrophic obstructive cardiomyopathy. Clinical cardiology. 2024 Feb:47(3):e24217. doi: 10.1002/clc.24217. Epub [PubMed PMID: 38439605]
Maron BJ, Rowin EJ, Maron MS. Hypertrophic Cardiomyopathy: New Concepts and Therapies. Annual review of medicine. 2022 Jan 27:73():363-375. doi: 10.1146/annurev-med-042220-021539. Epub [PubMed PMID: 35084989]
Tokodi M, Oláh A, Fábián A, Lakatos BK, Hizoh I, Ruppert M, Sayour AA, Barta BA, Kiss O, Sydó N, Csulak E, Ladányi Z, Merkely B, Kovács A, Radovits T. Novel insights into the athlete's heart: is myocardial work the new champion of systolic function? European heart journal. Cardiovascular Imaging. 2022 Jan 24:23(2):188-197. doi: 10.1093/ehjci/jeab162. Epub [PubMed PMID: 34432004]
Kübler J, Burgstahler C, Brendel JM, Gassenmaier S, Hagen F, Klingel K, Olthof SC, Blume K, Wolfarth B, Mueller KAL, Greulich S, Krumm P. Cardiac MRI findings to differentiate athlete's heart from hypertrophic (HCM), arrhythmogenic right ventricular (ARVC) and dilated (DCM) cardiomyopathy. The international journal of cardiovascular imaging. 2021 Aug:37(8):2501-2515. doi: 10.1007/s10554-021-02280-6. Epub 2021 May 21 [PubMed PMID: 34019206]