Introduction
Autosomal recessive polycystic kidney disease is a rare genetic disorder primarily affecting the kidneys and liver. Clinicians should recognize early signs such as enlarged, echogenic kidneys in utero or during infancy. ARPKD most commonly results from mutations in PKHD1, causing renal cysts and congenital hepatic fibrosis early in life. About half of the patients with ARPKD develop end-stage renal failure requiring renal replacement therapy (dialysis or kidney transplantation) within the first 2 decades of life. In addition, hepatic function must be closely followed, as portal hypertension, splenomegaly, and cholangitis are common, especially in older patients.
The 2 varieties of polycystic kidney disease (PKD) based on inheritance are the relatively common autosomal dominant and the much rarer autosomal recessive types.[1] These 2 forms have distinct clinical and genetic features.[1]
Autosomal dominant polycystic kidney disease (ADPKD), previously called adult polycystic kidney disease, is a multisystem progressive cystic disorder primarily affecting the kidneys. It is characterized by bilateral renal cysts, which progressively lead to fibrosis, architectural distortion, hypertension, and progressive renal failure, typically becoming symptomatic starting at about 30 years of age.[1][2][3] In adult patients, it is the most frequent genetic cause of renal failure and end-stage kidney disease. Please see the companion StatPearls reference, "Autosomal Dominant Polycystic Kidney Disease," for further information.[3]
Autosomal recessive polycystic kidney disease (ARPKD) primarily involves the kidney and liver. Historically referred to as infantile polycystic kidney disease, it can present in neonatal, infantile, juvenile, or even adult populations. Hence, the old nomenclature of adult and infantile polycystic kidney disease is not used anymore.[4]
ARPKD is characterized by renal distal tubule and collecting duct cyst formation, hepatic biliary duct ectasia/malformation, and fibrosis involving both the liver and kidneys.[5]
ARPKD is a ciliopathy and presents with cysts along the distal renal tubules and collecting ducts, compared to ADPKD, where the cysts can develop anywhere along the nephron. Cysts in ARPKD are mostly microcystic in early childhood, may develop calcifications, and can produce renal calculi. In older children, the cysts become larger and concentrated in the renal medulla.
The clinical presentation of ARPKD varies significantly, with some patients diagnosed at a young age having more severe symptoms than those diagnosed when older.[6] ARPKD is always associated with liver bile duct malformations, both intrahepatic and extrahepatic. Liver manifestations vary from mild cholestasis to hepatic fibrosis, portal vein hypertension, esophageal varices, cholangitis, and cirrhosis.[7]
A key factor in understanding the presentations of ARPKD is that 30% to 40% of affected neonates have been reported to die perinatally, usually due to respiratory distress from pulmonary hypoplasia from oligohydramnios related to decreased fetal urine production.[8][9] Improved neonatal care has reduced the newborn mortality rate to just 20%.[10]
Survival rates are 85% and 82% for those who survive the perinatal period at 1 and 10 years, respectively.[11][12] Of note, some of the perinatal data may have been derived from older studies conducted prior to technical advances in neonatal intensive care, reducing perinatal mortality to about 20% within the first month of life.[7]
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
There is considerable variability in the phenotypic expression of ARPKD. More than 90% of ARPKD cases are caused by mutations in the PKHD1 gene on chromosome 6p12.[13] Its function is to regulate the production of fibrocystin (polyductin), which is involved in cellular adhesion, ciliary function, and cell proliferation in the kidney and liver.[14][15][16]
There is evidence of extensive alternative splicing of this gene. A critical amount of full-length protein secreted by this gene is responsible for the normal function of tubular epithelium. More recently, DAZ-interacting zinc finger protein 1-like (DZIP1L) has been implicated in some cases of ARPKD, but it is extremely rare.[7]
Patients with tuberous sclerosis have mutations in TSC1 and TSC2. These genes are localized to chromosome 16, the same as PKD1. Patients with tuberous sclerosis often have an early onset of PKD and present similarly to ARPKD.
Epidemiology
ARPKD is one of the most common causes of heritable, infantile cystic renal disease. Despite that, it is found in only 1 in 20,000 to 40,000 live births.[17] In nonisolated populations, this suggests a carrier frequency of about 1 in 70.[5][10]
There is no gender, ethnic, or racial predilection. Patients who are heterozygous for ARPKD can show mild variations of polycystic kidney or hepatic cystic disease.[11] The number of children with early onset autosomal dominant polycystic kidney disease is comparable to the number with ARPKD, which can make differentiation difficult in the pediatric age group.[18][19]
Pathophysiology
The gene responsible for most ARPKD cases is Polycystic Kidney and Hepatic Disease 1 (PHKD1). The protein encoded by PKHD1 is fibrocystin (polyductin), a membrane protein found mainly in the distal tubules of the kidneys and biliary ducts of the liver.[20] The exact function of this protein is somewhat uncertain, but it is thought to be involved in cellular adhesion, ciliary function, and cell proliferation.[14][15][16]
Defects of PKHD1 also cause embryologic ductal plate malformations, leading to congenital hepatic fibrosis. Eventually, this results in variable dilatation in the intrahepatic and extrahepatic bile ducts (Caroli disease).[21] Associated clinical manifestations are primarily portal hypertension, splenomegaly, hepatic fibrosis, and cholangitis.[11][21] See the companion Statpearls reference, "Caroli Syndrome," for more information.[11][21]
Mammalian targets of rapamycin (mTOR) and cyclic AMP (cAMP) are 2 of these pathways being studied.[11] Animal ARPKD models testing epithelial growth receptor blockers and epithelial enzymes have shown promising results, which may pave the way for therapeutic interventions at the cellular level.[22]
The pathogenesis of ARPKD is characterized by circumferential proliferation of epithelial cells, which predominantly affects the collecting ducts in renal tubules.[23] The primary pathology involves abnormal epithelial cell proliferation, which forms cysts that detach from the tubule once they reach a certain size.[23] The abnormal proliferation of renal tubule epithelium causes them to lose their normal physiological function, and they fill with fluid from transepithelial secretion.[23]
The fluid is rich in epithelial growth factors, which leads to further proliferation of epithelial cells. This leads to extracellular matrix deposition and increased cytokines, growth factors, and inflammation. All these factors lead to renal fibrosis, proximal tubule apoptosis, and loss of kidney function.[11] Later in the disease, the kidney becomes massively enlarged with macrocysts and interstitial fibrosis— similar to the presentation of ADPKD.[11]
Defects of the PKHD1 gene also cause embryologic ductal plate malformation, leading to congenital hepatic fibrosis. Eventually, this results in variable dilatation in the intrahepatic and extrahepatic bile ducts (Caroli disease).[5][21] Associated clinical manifestations are primarily portal hypertension, splenomegaly, hepatic fibrosis, and cholangitis.[11]
Histopathology
Histology shows distal renal tubular and collecting duct cysts lined by columnar or cuboidal epithelium. A liver biopsy shows various degrees of diffuse fibrosis and dilated intrahepatic ducts.[5]
History and Physical
Early identification of patients with ARPKD has increased tremendously in the last few decades, with routine antenatal ultrasound usually showing very large, echogenic kidneys. Sometimes the kidneys are so enlarged they can interfere with normal vaginal delivery.
Perinatal demise for the first 30 days after birth is primarily due to respiratory distress from pulmonary hypoplasia and reduced vital capacity from thoracic compression by the markedly enlarged kidneys. Usually, in the first few months, infants will have renal dysfunction and hypertension (75%), which may be severe.
Failure to thrive and growth retardation is a significant concern, as nutritional status can be affected by excessively enlarged kidneys, portal hypertension, renal failure, and the possible need for peritoneal dialysis.
Symptoms that develop in early childhood include urinary tract infections, gross hematuria, and renal osteopathy. Neurocognitive function in children with ARPKD is not affected.[24]
Variable degrees of congenital hepatic fibrosis are present, which can lead to splenomegaly, thrombocytopenia, and esophageal varices with the potential for upper GI bleeding. Cholangitis is a common presentation and should be ruled out in any patient with ARPKD who presents with fever or abdominal pain.
Earlier (neonatal) diagnosis is associated with more severe kidney disease, while a later presentation tends to demonstrate milder degrees of renal impairment but more severe liver disease with portal hypertension.[25][26][27]
Physical examination findings may include Potter's facies, limb deformities, and other oligohydramnios-related signs. The kidneys are often markedly enlarged, and there may be splenomegaly. Potter's sequence may be present, including severe oligohydramnios, pulmonary hypoplasia, limb deformities from utero compression (eg, club feet, hip dislocations), and Potter's facies.)
Evaluation
ARPKD can present antenatally with evidence of oligohydramnios or enlarged fetal echogenic kidneys. Amniocentesis for gene karyotyping is an option if desired.[12] A detailed family history of renal disease or unexplained perinatal loss should be taken. The absence of renal findings in the patient's biological parents increases the suspicion of ARPKD and prompts consideration for further genetic testing.[28]
Laboratory testing should include kidney and liver function, electrolytes, serum albumin, coagulation studies, and a complete blood count, as thrombocytopenia from splenic sequestration is often seen, particularly with portal hypertension. Platelet count and thrombocytopenia are excellent indicators of the severity of portal hypertension (frequently underdiagnosed) in patients with ARPKD.[26]
Neonates may not be able to produce dilute urine and may, therefore, demonstrate hyponatremia for the first few weeks immediately after birth.[29] Be aware that serum laboratory tests on newborns (eg, electrolytes and creatinine) will reflect maternal serum values for the first 24 to 48 hours after birth.
Ultrasonography is the preferred imaging modality, particularly in the perinatal and neonatal periods.[30][31] Antenatal diagnosis can be made by the 24th week of gestation, although it has occasionally been identified as early as 18 weeks after conception. Ultrasound typically shows bilateral smooth, massively enlarged kidneys with loss of corticomedullary differentiation. Increased echogenicity and microcysts are typical and common.[30] The markedly enlarged kidneys may concentrate the bowel loops towards the midline.
In older children with ARPKD, ultrasound may show larger cysts and increased echogenicity from cysts arising from the distal tubules and collecting ducts, which are concentrated in the renal medulla. Contrast-enhanced ultrasound can be performed but requires additional expertise.[30]
Unlike ADPKD, total kidney volume does not necessarily correlate with renal function.[12] Hepatic ultrasound may demonstrate hepatomegaly, increased echogenicity, hepatic cysts, and dilatation of peripheral parenchymal and extrahepatic biliary ducts.[31] Biliary abnormalities will be found at some point in 70% of ARPKD patients.[26]
Magnetic resonance imaging (MRI) can be used instead as it avoids ionizing radiation and shows hepatic lesions well, but this often requires sedation, which carries its risks.[30] Cysts are best imaged on T2-weighted sequences.[30] CT and MRI scans are not necessary for a diagnosis of ARPKD but may be helpful in selected cases. CT scans should be avoided when possible in children due to the radiation exposure.
In older patients, CT imaging shows smooth, enlarged kidneys with a striated pattern of contrast excretion.[32] The striated pattern on CT signifies a collection of contrast in dilated renal collecting tubules.
The diagnosis of ARPKD is made in most cases by a combination of renal ultrasound findings, clinical presentation, and the lack of a family history of kidney disease.[33][34]
Genetic testing is beneficial and recommended in confirming a diagnosis of ARPKD, especially if the history, clinical findings, and imaging are inconclusive, as most patients will have an identifiable gene mutation (PHKD1).[19][35][36][37][38][39][40] While genetic testing is considered the "gold standard" diagnostic modality, it may miss some cases due to the large number of genetic variants.[39][40] Overall reliability (positive predictive value) of genetic testing is reported at 80% to 85%.[8][9][39][41]
A definitive diagnosis confirmed by genetic testing clearly establishes a prognosis, improves clinical management, facilitates the early detection of comorbidities and complications, avoids unnecessary procedures and invasive testing, optimizes the timing and focus of follow-up evaluations, and allows proper genetic counseling for the patient and family.[19]
Once a diagnosis of ARPKD is made, involvement of the liver should be assumed. The initial hepatic ultrasound is often routine or may show only minimal cysts. A baseline hepatic MRI can be considered. Liver chemistries are usually normal in neonates, as hepatic function is generally preserved early in life. Liver function can be followed by laboratory testing (liver enzymes, bilirubin, coagulation studies, serum albumin, and platelet count), periodic hepatic ultrasound, and MRI imaging.
The first signs of congenital hepatic fibrosis are usually portal hypertension and splenomegaly, which may manifest as thrombocytopenia or gastrointestinal (GI) bleeding.[12]
Consensus guidelines suggest further surveillance imaging as follows:
- Liver and spleen ultrasound with Doppler of the portal and splenic veins monitoring for intrahepatic or extrahepatic biliary dilatation.
- Annual laboratory tests for complete blood counts (especially platelets) should be followed to assess for splenomegaly.
- A complete abdominal ultrasound should be performed on children aged 5 years with special attention to the bile ducts and spleen size. Any abnormalities should prompt referral to a pediatric hepatologist and screening for complications following the standard guidelines.[12]
Up to 5% of patients with ADPKD present in childhood, prenatally, or as neonates, making the incidence of the 2 polycystic kidney disorders comparable.[11][19] Differentiating ARPKD from ADPKD can sometimes be challenging as the clinical presentations overlap.[3][33]
ARPKD is a more severe form of the disorder that typically appears early, usually at birth, and can often be diagnosed antenatally. The facts to consider regarding ARPKD are as follows:
- The condition is relatively rare at 1:20,000 to 40,000 live births.[33]
- Cerebral aneurysms are extremely rare.
- No specific cardiovascular anomalies are observed.
- Liver disease is always present and likely to eventually become significant or severe.[33]
- No family history of renal cystic disease or kidney failure is usually present.
- Perinatal mortality is high, and end-stage kidney disease affects half of those who survive to age 20.[33]
ADPKD disease is a slower and milder form of the condition that usually demonstrates symptoms starting at about age 30.[3] The facts to consider regarding ADPKD are as follows:
- The disorder is relatively common at 1:400 to 1:1,000 live births.[3]
- Cerebral aneurysms may be present.
- Cardiac anomalies may be present, especially mitral valve prolapse.
- Liver cysts may be present, but not usually early, and overall liver function is not usually affected.[3]
- A family history of hypertension and kidney disease is very likely.
- Overall life expectancy with proper treatment averages up to age 70.
- End-stage kidney disease develops in most patients starting in their 60's.[3]
Treatment / Management
Perinatal management depends on the severity of the clinical manifestations and the organs involved. This involves monitoring respiratory function, renal function tests, liver function tests, infant growth evaluation, blood pressure monitoring, and symptomatic treatment. Cesarean delivery may be preferred if ARPKD is diagnosed antenatally due to the increased risk of dystocia from the massively enlarged fetal kidneys.[39][42][43] (B3)
Nephrectomy is occasionally required for significantly enlarged kidneys compromising respiratory or gastrointestinal function but is associated with significant complications.[12] ARPKD patients with end-stage kidney disease who ultimately receive renal transplants can benefit from nephrectomies of their nonfunctional kidneys by making more room available for the new kidney and facilitating blood pressure control after transplantation.[5] (B3)
Initial treatment may consist of respiratory support, fluid and electrolyte management (for hyponatremia or hyperkalemia), control of hypertension, nutritional support, bicarbonate or citrate supplements for metabolic acidosis, antibiotics for infections, and peritoneal dialysis if required.[5][7]
Pulmonary hypoplasia can often be managed by respiratory supportive measures such as mechanical ventilation, allowing the lungs to mature further. Pulmonary hypertension can sometimes be treated with nitrous oxide, and the use of palivizumab, a prophylactic monoclonal antibody for respiratory syncytial virus, is recommended.[33] Almost half of ARPKD patients diagnosed prenatally will be born with clinically significant pulmonary hypoplasia. Respiratory problems are the leading cause of death in neonates with ARPKD.
Significant renal failure may induce hyperkalemia, while hyponatremia is often found in neonates with ARPKD. The hyponatremia is usually transient and treated with fluid restriction.[44] Patients with liver disease may need supplemental bile acids, endoscopic control of varices, or a portal vein shunting procedure. Renal replacement therapy, if indicated, can include hemodialysis or renal transplantation, but usually, peritoneal dialysis is preferred. Some patients may need nephrectomies to allow sufficient space for adequate peritoneal dialysis.(B3)
Patients with significant renal failure may also require iron and erythropoietin therapy for the management of anemia, calcium and vitamin D supplements for bone health, phosphate binders for hyperphosphatemia, sodium bicarbonate or potassium citrate for metabolic acidosis, and medical parathyroid hormone suppression with a calcimimetic agent. The use of recombinant human growth hormone appears to be safe and effective in promoting normal growth, which might otherwise be suppressed by uremia and reduced nutritional intake.[45]
ARPKD patients who eventually receive renal transplants are at higher risk of death from cholangitis and biliary sepsis due to the required immunosuppressive drugs, which is a concern.[46]
Hypertension is often severe, requiring multiple medications for control, starting with renin-angiotensin-aldosterone system (RAAS) blockers, which are usually the first-line treatments.[7] Beta-blockers, calcium channel blockers, dietary salt restriction, and diuretics can be used to complement RAAS inhibitors, when necessary, to achieve adequate blood pressure control. The appearance of hypertension generally precedes any clinical evidence of renal failure and will be present in 80% of children with ARPKD.[9] Management of hypertension is usually easier after the first year of life.[5]
The ESCAPE trial (published in 2009) noted that children with stages 2 to 4 chronic kidney disease from all causes had improved renal function with aggressive blood pressure control.[12](B3)
Nutritional support can be highly beneficial in patients with ARPKD, particularly during the first two years after birth.[33][47] This is especially important in those children with significant portal hypertension and early renal failure.[33][47] The poor feeding is primarily due to compression on the stomach and GI tract by the extremely enlarged polycystic kidneys. The use of recombinant human growth hormone has been considered to promote growth and appears safe and effective in limited studies.[45]
Liver disease requires rapid treatment of ascending cholangitis and may benefit from bile acid supplements, such as ursodeoxycholic acid, to help increase the natural hepatic biliary secretions and help minimize the development of cholelithiasis.[33] Endoscopic sclerotherapy or banding of esophageal varices may be needed in patients with progressive portal hypertension.[33] In more severe cases, portal vein shunting or a liver transplant may need to be considered. Prophylactic antibiotics to help prevent ascending cholangitis are recommended by some experts.[35]
Dual organ transplants (liver and kidney), depending on the severity of portal hypertension and end-stage renal disease, have shown promising results in a significant number of cases and may be a consideration in selected patients.[47]
Currently, tolvaptan, a vasopressin receptor antagonist, is the only FDA-approved treatment for ADPKD, and several large trials with long-term follow-up have demonstrated efficacy in preserving renal function.[3][48][49][50][51] Ongoing clinical trials are studying tolvaptan for the possible treatment of ARPKD.[35] Other possible therapies are under investigation.(A1)
Differential Diagnosis
The differential diagnosis for ARPKD is broad and includes the following:
- ADPKD [3]
- Antenatal cytomegalovirus (CMV) infections can cause increased echogenicity of fetal kidneys on ultrasound. This can be tested by amniocentesis.[12]
- Autosomal dominant tubulointerstitial kidney disease, also called medullary cystic kidney disease, is an autosomal dominant disorder with a similar presentation to nephronophthisis (see below) but presents at a later age.[52] Interstitial and tubular fibrosis are prevalent, and several different gene mutations are associated with this condition.[52][53]
- Bardet–Biedl, Joubert, and Meckel syndromes are also ciliopathies with presentations similar to ARPKD.[19][54][55]
- Bardet–Biedl syndrome often presents with enlarged and hyperechogenic kidneys with a loss of corticomedullary differentiation.
- Joubert and Meckel syndromes are more severe ciliopathies characterized by neurological problems and delayed development, as well as possible liver fibrosis, polydactyly, and cystic kidneys.
- Hereditary renal agenesis or dysplasia. One series found that patients with renal agenesis or dysplasia are much more likely to have first-degree relatives with renal malformations.[56][57]
- Hepatocyte nuclear factor 1-beta (HNF1β) is a transcription factor gene regulating the expression of PKHD1, and mutations here can cause various clinical symptoms, including renal cysts mimicking ARPKD.[58]
- Nephronophthisis is another autosomal recessive cystic kidney disease manifesting in tubulointerstitial microcysts and renal fibrosis.[59] Nephronophthisis commonly causes end-stage renal disease (ESRD) in patients younger than 25.[1][59]
- Von Hippel–Lindau syndrome is an autosomal dominant disease caused by mutations in the tumor suppressor VHL gene.[60] Primary symptoms are hemangioblastomas in the central nervous system and renal tumors.[60] Affected people also have a high probability of renal and pancreatic cysts. The similarity in manifestations between tuberous sclerosis, von Hippel Landau, and polycystic kidney disease suggests a functional connection involving primary cilium and the mTOR signaling pathway.[60]
Prognosis
The prognosis depends on the severity of the pulmonary, hepatic, and renal disease. Neonates born with severe renal disease may not survive due to pulmonary hypoplasia and insufficiency, which may occur in up to 40% of cases.[8][9]
Neonatal mortality has improved due to advances in neonatal care but remains at about 20%.[10] Those who survive the neonatal period have 10-year survival rates estimated at 82% but often develop progressive renal failure, hypertension, hepatic fibrosis, portal hypertension, and end-stage renal disease.[9][39] Other reports suggest that over 90% of those who survive the neonatal period will survive to age 20.
The liver disease in ARPKD is relatively mild in neonates and early childhood, but the severity of the hepatic dysfunction progresses with age. These children develop features of portal hypertension because of chronic hepatic fibrosis. However, as portal hypertension and variceal bleeding are not generally life-threatening if properly managed, many of these patients survive up to middle age. Patients with ARPKD older than 40 have a slightly increased risk of hepatic tumors, especially cholangiocarcinoma.[11]
Complications
Complications associated with ARPKD include the following:
- Azotemia
- Cholangitis
- Congenital hepatic fibrosis
- Gastrointestinal bleeding
- Hypertension, often severe
- Increased risk for ascending cholangitis
- Large bilateral flank masses
- Liver failure and cirrhosis
- Malabsorption of fat-soluble vitamins
- Poor growth, feeding, and nutrition
- Progressive portal hypertension with esophageal and gastric varices, splenomegaly, and GI bleeds
- Progressive renal failure
- Renal osteopathy
- Respiratory distress due to pulmonary hypoplasia and limited diaphragmatic mobility due to the massively enlarged kidneys
- Splenomegaly (present in about 60% of patients)
- Thrombocytopenia
Deterrence and Patient Education
The risk to parents of having another baby with ARPKD after having had one child with the disorder is 25% due to its autosomal recessive inheritance pattern.[19] With gene penetration at 1:70, the estimated risk of ARPKD is as follows:
- Children of patients with ARPKD: 1:140.
- Children of healthy siblings: 1:420.
- Children of healthy aunts and uncles: 1:560.[19]
Any diagnosis of ARPKD should prompt evaluation in first-degree relatives, especially siblings who have a 66% risk of being a carrier for ARPKD, even if apparently healthy.[19] Patients with ARPKD or a family history of this genetic disorder should be made aware of the risk in future children, and amniocentesis can be offered to patients who request this service.
Patients and their families must receive education regarding their condition, likely disease progression, complications, and management options, as well as genetic testing and appropriate counseling. Contact sports are not recommended for patients with ARPKD. Social support to patients and families during treatment and management is paramount.
Pearls and Other Issues
Key facts to keep in mind regarding ARPKD include the following:
- Renal ultrasounds of both parents can be helpful and are suggested as well as in siblings.
- Genetic testing should be considered in siblings and other appropriate family members so proper genetic counseling can be made available. Such counseling should be involved in all cases where ARPKD has been diagnosed.
- Although ARPKD and ADPKD may seem far apart clinically, they can overlap. In those cases, evidence of hepatic involvement, family history, and particularly genetic testing are the best ways to differentiate them.
Enhancing Healthcare Team Outcomes
Patients with ARPKD are at high risk of ESRD. Early identification and management of patients with ARPKD are imperative in reducing morbidity and mortality. The care of patients with ARPKD can begin antenatally with the detection of oligohydramnios or abnormalities of the renal system. Initial complications include pulmonary hypoplasia, which may necessitate mechanical ventilation and monitoring in a neonatal intensive care unit. Liver involvement is always present and frequently becomes symptomatic as patients mature.
ARPKD necessitates a collaborative approach among healthcare professionals to ensure patient-centered care and improve overall outcomes. Obstetricians, maternal-fetal medicine specialists, pediatric nephrologists, adult nephrologists, urologists, pediatric urologists, critical care physicians, gastroenterologists, hepatologists, advanced practitioners, nurses, pharmacists, and the many other health professionals involved in the care of these complex patients should possess the essential clinical skills and knowledge to diagnose and manage ARPKD.
This includes expertise in recognizing the varied clinical presentations and understanding the nuances of diagnostic techniques such as renal ultrasound, MRI, and abdominal imaging. Patient and caregiver education about the genetic component of ARPKD, the natural progression of associated renal disease, and common renal and gastrointestinal symptoms are essential to prevent morbidity.
Effective interprofessional communication is paramount, allowing seamless information exchange and collaborative decision-making among the team members. Care coordination plays a pivotal role in ensuring that the patient's journey from diagnosis to treatment and follow-up is well-managed, minimizing errors and enhancing patient safety. By embracing these principles of skill, strategy, interprofessional communication, and care coordination, healthcare professionals can deliver optimal patient-centered care, ultimately improving patient outcomes and enhancing team performance in the management of ARPKD.
Media
(Click Image to Enlarge)
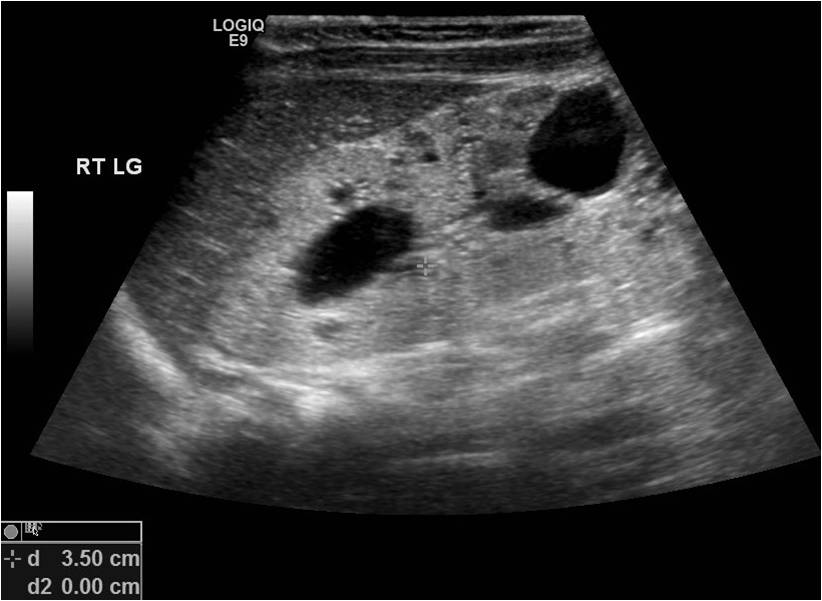
ARPKD in a 6-month -old girl. Enalrged and ecogenic kidney with cysts. Contributed by Surabhi Subramanian .
(Click Image to Enlarge)
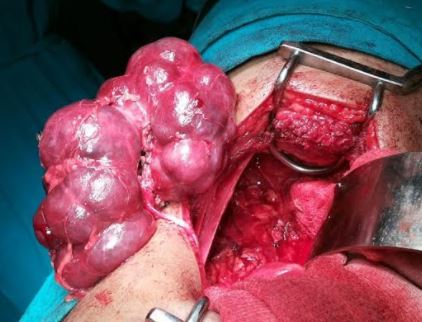
Polycystic kidney disease Contributed by Sunil Munakomi, MD
References
Goksu SY, Leslie SW, Khattar D. Renal Cystic Disease. StatPearls. 2024 Jan:(): [PubMed PMID: 32119391]
Torres VE, Harris PC. Autosomal dominant polycystic kidney disease: the last 3 years. Kidney international. 2009 Jul:76(2):149-68. doi: 10.1038/ki.2009.128. Epub 2009 May 20 [PubMed PMID: 19455193]
Mahboob M, Rout P, Bokhari SRA. Autosomal Dominant Polycystic Kidney Disease. StatPearls. 2024 Jan:(): [PubMed PMID: 30422529]
Guay-Woodford LM, Galliani CA, Musulman-Mroczek E, Spear GS, Guillot AP, Bernstein J. Diffuse renal cystic disease in children: morphologic and genetic correlations. Pediatric nephrology (Berlin, Germany). 1998 Apr:12(3):173-82 [PubMed PMID: 9630032]
Bergmann C, Guay-Woodford LM, Harris PC, Horie S, Peters DJM, Torres VE. Polycystic kidney disease. Nature reviews. Disease primers. 2018 Dec 6:4(1):50. doi: 10.1038/s41572-018-0047-y. Epub 2018 Dec 6 [PubMed PMID: 30523303]
Goggolidou P, Richards T. The genetics of Autosomal Recessive Polycystic Kidney Disease (ARPKD). Biochimica et biophysica acta. Molecular basis of disease. 2022 Apr 1:1868(4):166348. doi: 10.1016/j.bbadis.2022.166348. Epub 2022 Jan 12 [PubMed PMID: 35032595]
Liebau MC. Early clinical management of autosomal recessive polycystic kidney disease. Pediatric nephrology (Berlin, Germany). 2021 Nov:36(11):3561-3570. doi: 10.1007/s00467-021-04970-8. Epub 2021 Feb 17 [PubMed PMID: 33594464]
Guay-Woodford LM, Desmond RA. Autosomal recessive polycystic kidney disease: the clinical experience in North America. Pediatrics. 2003 May:111(5 Pt 1):1072-80 [PubMed PMID: 12728091]
Level 3 (low-level) evidenceBergmann C, Senderek J, Windelen E, Küpper F, Middeldorf I, Schneider F, Dornia C, Rudnik-Schöneborn S, Konrad M, Schmitt CP, Seeman T, Neuhaus TJ, Vester U, Kirfel J, Büttner R, Zerres K, APN (Arbeitsgemeinschaft für Pädiatrische Nephrologie). Clinical consequences of PKHD1 mutations in 164 patients with autosomal-recessive polycystic kidney disease (ARPKD). Kidney international. 2005 Mar:67(3):829-48 [PubMed PMID: 15698423]
Alzarka B, Morizono H, Bollman JW, Kim D, Guay-Woodford LM. Design and Implementation of the Hepatorenal Fibrocystic Disease Core Center Clinical Database: A Centralized Resource for Characterizing Autosomal Recessive Polycystic Kidney Disease and Other Hepatorenal Fibrocystic Diseases. Frontiers in pediatrics. 2017:5():80. doi: 10.3389/fped.2017.00080. Epub 2017 Apr 20 [PubMed PMID: 28473971]
Cordido A, Vizoso-Gonzalez M, Garcia-Gonzalez MA. Molecular Pathophysiology of Autosomal Recessive Polycystic Kidney Disease. International journal of molecular sciences. 2021 Jun 17:22(12):. doi: 10.3390/ijms22126523. Epub 2021 Jun 17 [PubMed PMID: 34204582]
Guay-Woodford LM, Bissler JJ, Braun MC, Bockenhauer D, Cadnapaphornchai MA, Dell KM, Kerecuk L, Liebau MC, Alonso-Peclet MH, Shneider B, Emre S, Heller T, Kamath BM, Murray KF, Moise K, Eichenwald EE, Evans J, Keller RL, Wilkins-Haug L, Bergmann C, Gunay-Aygun M, Hooper SR, Hardy KK, Hartung EA, Streisand R, Perrone R, Moxey-Mims M. Consensus expert recommendations for the diagnosis and management of autosomal recessive polycystic kidney disease: report of an international conference. The Journal of pediatrics. 2014 Sep:165(3):611-7. doi: 10.1016/j.jpeds.2014.06.015. Epub 2014 Jul 9 [PubMed PMID: 25015577]
Level 3 (low-level) evidenceBergmann C, Senderek J, Küpper F, Schneider F, Dornia C, Windelen E, Eggermann T, Rudnik-Schöneborn S, Kirfel J, Furu L, Onuchic LF, Rossetti S, Harris PC, Somlo S, Guay-Woodford L, Germino GG, Moser M, Büttner R, Zerres K. PKHD1 mutations in autosomal recessive polycystic kidney disease (ARPKD). Human mutation. 2004 May:23(5):453-63 [PubMed PMID: 15108277]
Ward CJ, Hogan MC, Rossetti S, Walker D, Sneddon T, Wang X, Kubly V, Cunningham JM, Bacallao R, Ishibashi M, Milliner DS, Torres VE, Harris PC. The gene mutated in autosomal recessive polycystic kidney disease encodes a large, receptor-like protein. Nature genetics. 2002 Mar:30(3):259-69 [PubMed PMID: 11919560]
Level 3 (low-level) evidenceOnuchic LF, Furu L, Nagasawa Y, Hou X, Eggermann T, Ren Z, Bergmann C, Senderek J, Esquivel E, Zeltner R, Rudnik-Schöneborn S, Mrug M, Sweeney W, Avner ED, Zerres K, Guay-Woodford LM, Somlo S, Germino GG. PKHD1, the polycystic kidney and hepatic disease 1 gene, encodes a novel large protein containing multiple immunoglobulin-like plexin-transcription-factor domains and parallel beta-helix 1 repeats. American journal of human genetics. 2002 May:70(5):1305-17 [PubMed PMID: 11898128]
Menezes LF, Cai Y, Nagasawa Y, Silva AM, Watkins ML, Da Silva AM, Somlo S, Guay-Woodford LM, Germino GG, Onuchic LF. Polyductin, the PKHD1 gene product, comprises isoforms expressed in plasma membrane, primary cilium, and cytoplasm. Kidney international. 2004 Oct:66(4):1345-55 [PubMed PMID: 15458427]
Level 3 (low-level) evidenceTraubici J, Daneman A. High-resolution renal sonography in children with autosomal recessive polycystic kidney disease. AJR. American journal of roentgenology. 2005 May:184(5):1630-3 [PubMed PMID: 15855129]
Bergmann C, Zerres K. Early manifestations of polycystic kidney disease. Lancet (London, England). 2007 Jun 30:369(9580):2157. doi: 10.1016/S0140-6736(07)61005-8. Epub [PubMed PMID: 17604790]
Bergmann C. Genetics of Autosomal Recessive Polycystic Kidney Disease and Its Differential Diagnoses. Frontiers in pediatrics. 2017:5():221. doi: 10.3389/fped.2017.00221. Epub 2018 Feb 9 [PubMed PMID: 29479522]
Ward CJ, Yuan D, Masyuk TV, Wang X, Punyashthiti R, Whelan S, Bacallao R, Torra R, LaRusso NF, Torres VE, Harris PC. Cellular and subcellular localization of the ARPKD protein; fibrocystin is expressed on primary cilia. Human molecular genetics. 2003 Oct 15:12(20):2703-10 [PubMed PMID: 12925574]
Umar J, Kudaravalli P, John S. Caroli Disease. StatPearls. 2024 Jan:(): [PubMed PMID: 30020679]
Richards WG, Sweeney WE, Yoder BK, Wilkinson JE, Woychik RP, Avner ED. Epidermal growth factor receptor activity mediates renal cyst formation in polycystic kidney disease. The Journal of clinical investigation. 1998 Mar 1:101(5):935-9 [PubMed PMID: 9486961]
Level 3 (low-level) evidenceMartinez JR, Grantham JJ. Polycystic kidney disease: etiology, pathogenesis, and treatment. Disease-a-month : DM. 1995 Nov:41(11):693-765 [PubMed PMID: 7587886]
Level 3 (low-level) evidenceHartung EA, Matheson M, Lande MB, Dell KM, Guay-Woodford LM, Gerson AC, Warady BA, Hooper SR, Furth SL. Neurocognition in children with autosomal recessive polycystic kidney disease in the CKiD cohort study. Pediatric nephrology (Berlin, Germany). 2014 Oct:29(10):1957-65. doi: 10.1007/s00467-014-2816-5. Epub 2014 May 15 [PubMed PMID: 24828609]
Level 2 (mid-level) evidenceShaikewitz ST, Chapman A. Autosomal recessive polycystic kidney disease: issues regarding the variability of clinical presentation. Journal of the American Society of Nephrology : JASN. 1993 Jun:3(12):1858-62 [PubMed PMID: 8338916]
Gunay-Aygun M, Font-Montgomery E, Lukose L, Tuchman Gerstein M, Piwnica-Worms K, Choyke P, Daryanani KT, Turkbey B, Fischer R, Bernardini I, Sincan M, Zhao X, Sandler NG, Roque A, Douek DC, Graf J, Huizing M, Bryant JC, Mohan P, Gahl WA, Heller T. Characteristics of congenital hepatic fibrosis in a large cohort of patients with autosomal recessive polycystic kidney disease. Gastroenterology. 2013 Jan:144(1):112-121.e2. doi: 10.1053/j.gastro.2012.09.056. Epub 2012 Oct 3 [PubMed PMID: 23041322]
Zerres K, Rudnik-Schöneborn S, Steinkamm C, Becker J, Mücher G. Autosomal recessive polycystic kidney disease. Journal of molecular medicine (Berlin, Germany). 1998 Apr:76(5):303-9 [PubMed PMID: 9587064]
Sweeney WE Jr, Avner ED. Pathophysiology of childhood polycystic kidney diseases: new insights into disease-specific therapy. Pediatric research. 2014 Jan:75(1-2):148-57. doi: 10.1038/pr.2013.191. Epub 2013 Oct 31 [PubMed PMID: 24336431]
Level 3 (low-level) evidenceKaplan BS, Fay J, Shah V, Dillon MJ, Barratt TM. Autosomal recessive polycystic kidney disease. Pediatric nephrology (Berlin, Germany). 1989 Jan:3(1):43-9 [PubMed PMID: 2702087]
Gimpel C, Avni EF, Breysem L, Burgmaier K, Caroli A, Cetiner M, Haffner D, Hartung EA, Franke D, König J, Liebau MC, Mekahli D, Ong ACM, Pape L, Titieni A, Torra R, Winyard PJD, Schaefer F. Imaging of Kidney Cysts and Cystic Kidney Diseases in Children: An International Working Group Consensus Statement. Radiology. 2019 Mar:290(3):769-782. doi: 10.1148/radiol.2018181243. Epub 2019 Jan 1 [PubMed PMID: 30599104]
Level 3 (low-level) evidenceTurkbey B, Ocak I, Daryanani K, Font-Montgomery E, Lukose L, Bryant J, Tuchman M, Mohan P, Heller T, Gahl WA, Choyke PL, Gunay-Aygun M. Autosomal recessive polycystic kidney disease and congenital hepatic fibrosis (ARPKD/CHF). Pediatric radiology. 2009 Feb:39(2):100-11. doi: 10.1007/s00247-008-1064-x. Epub 2008 Dec 17 [PubMed PMID: 19089418]
Gleason DC, McAlister WH, Kissane J. Cystic disease of the kidneys in children. The American journal of roentgenology, radium therapy, and nuclear medicine. 1967 May:100(1):135-46 [PubMed PMID: 4960727]
Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, Burgmaier K, Gimpel C, Schaefer F, Liebau M. Autosomal Recessive Polycystic Kidney Disease – PKHD1. GeneReviews(®). 1993:(): [PubMed PMID: 20301501]
Hoyer PF. Clinical manifestations of autosomal recessive polycystic kidney disease. Current opinion in pediatrics. 2015 Apr:27(2):186-92. doi: 10.1097/MOP.0000000000000196. Epub [PubMed PMID: 25689455]
Level 3 (low-level) evidenceMekahli D, Liebau MC, Cadnapaphornchai MA, Goldstein SL, Greenbaum LA, Litwin M, Seeman T, Schaefer F, Guay-Woodford LM. Design of two ongoing clinical trials of tolvaptan in the treatment of pediatric patients with autosomal recessive polycystic kidney disease. BMC nephrology. 2023 Feb 13:24(1):33. doi: 10.1186/s12882-023-03072-x. Epub 2023 Feb 13 [PubMed PMID: 36782137]
Edrees BM, Athar M, Al-Allaf FA, Taher MM, Khan W, Bouazzaoui A, Al-Harbi N, Safar R, Al-Edressi H, Alansary K, Anazi A, Altayeb N, Ahmed MA, Abduljaleel Z. Next-generation sequencing for molecular diagnosis of autosomal recessive polycystic kidney disease. Gene. 2016 Oct 10:591(1):214-226. doi: 10.1016/j.gene.2016.07.021. Epub 2016 Jul 9 [PubMed PMID: 27401137]
Melchionda S, Palladino T, Castellana S, Giordano M, Benetti E, De Bonis P, Zelante L, Bisceglia L. Expanding the mutation spectrum in 130 probands with ARPKD: identification of 62 novel PKHD1 mutations by sanger sequencing and MLPA analysis. Journal of human genetics. 2016 Sep:61(9):811-21. doi: 10.1038/jhg.2016.58. Epub 2016 May 26 [PubMed PMID: 27225849]
Yang H, Sieben CJ, Schauer RS, Harris PC. Genetic Spectrum of Polycystic Kidney and Liver Diseases and the Resulting Phenotypes. Advances in kidney disease and health. 2023 Sep:30(5):397-406. doi: 10.1053/j.akdh.2023.04.004. Epub [PubMed PMID: 38097330]
Level 3 (low-level) evidenceSociety for Maternal-Fetal Medicine (SMFM), Swanson K. Autosomal recessive polycystic kidney disease. American journal of obstetrics and gynecology. 2021 Nov:225(5):B7-B8. doi: 10.1016/j.ajog.2021.06.038. Epub 2021 Sep 8 [PubMed PMID: 34507795]
Wicher D, Obrycki Ł, Jankowska I. Autosomal Recessive Polycystic Kidney Disease-The Clinical Aspects and Diagnostic Challenges. Journal of pediatric genetics. 2021 Mar:10(1):1-8. doi: 10.1055/s-0040-1714701. Epub 2020 Jul 29 [PubMed PMID: 33552631]
Bergmann C, Senderek J, Schneider F, Dornia C, Küpper F, Eggermann T, Rudnik-Schöneborn S, Kirfel J, Moser M, Büttner R, Zerres K. PKHD1 mutations in families requesting prenatal diagnosis for autosomal recessive polycystic kidney disease (ARPKD). Human mutation. 2004 May:23(5):487-95 [PubMed PMID: 15108281]
Dukic L, Schaffelder R, Schaible T, Sütterlin M, Siemer J. [Massive increase of foetal abdominal circumference due to hereditary polycystic kidney disease]. Zeitschrift fur Geburtshilfe und Neonatologie. 2010 Jun:214(3):119-22. doi: 10.1055/s-0029-1225641. Epub 2010 Jun 23 [PubMed PMID: 20574939]
Belin S, Delco C, Parvex P, Hanquinet S, Fokstuen S, Martinez de Tejada B, Eperon I. Management of delivery of a fetus with autosomal recessive polycystic kidney disease: a case report of abdominal dystocia and review of the literature. Journal of medical case reports. 2019 Dec 12:13(1):366. doi: 10.1186/s13256-019-2293-3. Epub 2019 Dec 12 [PubMed PMID: 31829256]
Level 3 (low-level) evidenceHartung EA, Guay-Woodford LM. Autosomal recessive polycystic kidney disease: a hepatorenal fibrocystic disorder with pleiotropic effects. Pediatrics. 2014 Sep:134(3):e833-45. doi: 10.1542/peds.2013-3646. Epub 2014 Aug 11 [PubMed PMID: 25113295]
Level 3 (low-level) evidenceLilova M, Kaplan BS, Meyers KE. Recombinant human growth hormone therapy in autosomal recessive polycystic kidney disease. Pediatric nephrology (Berlin, Germany). 2003 Jan:18(1):57-61 [PubMed PMID: 12488992]
Davis ID, Ho M, Hupertz V, Avner ED. Survival of childhood polycystic kidney disease following renal transplantation: the impact of advanced hepatobiliary disease. Pediatric transplantation. 2003 Oct:7(5):364-9 [PubMed PMID: 14738296]
Telega G, Cronin D, Avner ED. New approaches to the autosomal recessive polycystic kidney disease patient with dual kidney-liver complications. Pediatric transplantation. 2013 Jun:17(4):328-35. doi: 10.1111/petr.12076. Epub 2013 Apr 17 [PubMed PMID: 23593929]
Müller RU, Messchendorp AL, Birn H, Capasso G, Cornec-Le Gall E, Devuyst O, van Eerde A, Guirchoun P, Harris T, Hoorn EJ, Knoers NVAM, Korst U, Mekahli D, Le Meur Y, Nijenhuis T, Ong ACM, Sayer JA, Schaefer F, Servais A, Tesar V, Torra R, Walsh SB, Gansevoort RT. An update on the use of tolvaptan for autosomal dominant polycystic kidney disease: consensus statement on behalf of the ERA Working Group on Inherited Kidney Disorders, the European Rare Kidney Disease Reference Network and Polycystic Kidney Disease International. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association. 2022 Apr 25:37(5):825-839. doi: 10.1093/ndt/gfab312. Epub [PubMed PMID: 35134221]
Level 3 (low-level) evidenceLacquaniti A. [Tolvaptan and autosomal dominant polycystic kidney disease in the adult: let's give time to the "TEMPO" trial (Tolvaptan Efficacy and Safety in Management of Autosomal Dominant Polycystic Kidney Disease and Its Outcomes)]. Giornale italiano di nefrologia : organo ufficiale della Societa italiana di nefrologia. 2013 Jan-Feb:30(1):. pii: gin/30.1.10. Epub [PubMed PMID: 25083527]
Torres VE, Higashihara E, Devuyst O, Chapman AB, Gansevoort RT, Grantham JJ, Perrone RD, Ouyang J, Blais JD, Czerwiec FS, TEMPO 3:4 Trial Investigators. Effect of Tolvaptan in Autosomal Dominant Polycystic Kidney Disease by CKD Stage: Results from the TEMPO 3:4 Trial. Clinical journal of the American Society of Nephrology : CJASN. 2016 May 6:11(5):803-811. doi: 10.2215/CJN.06300615. Epub 2016 Feb 23 [PubMed PMID: 26912543]
Chebib FT, Zhou X, Garbinsky D, Davenport E, Nunna S, Oberdhan D, Fernandes A. Tolvaptan and Kidney Function Decline in Older Individuals With Autosomal Dominant Polycystic Kidney Disease: A Pooled Analysis of Randomized Clinical Trials and Observational Studies. Kidney medicine. 2023 Jun:5(6):100639. doi: 10.1016/j.xkme.2023.100639. Epub 2023 Apr 14 [PubMed PMID: 37250503]
Level 1 (high-level) evidenceShamam YM, Hashmi MF. Autosomal Dominant Tubulointerstitial Kidney Disease. StatPearls. 2024 Jan:(): [PubMed PMID: 33760469]
Garfield K, Leslie SW. Medullary Sponge Kidney. StatPearls. 2024 Jan:(): [PubMed PMID: 29262095]
Van De Weghe JC, Gomez A, Doherty D. The Joubert-Meckel-Nephronophthisis Spectrum of Ciliopathies. Annual review of genomics and human genetics. 2022 Aug 31:23():301-329. doi: 10.1146/annurev-genom-121321-093528. Epub 2022 Jun 2 [PubMed PMID: 35655331]
Spahiu L, Behluli E, Grajçevci-Uka V, Liehr T, Temaj G. Joubert syndrome: Molecular basis and treatment. Journal of mother and child. 2022 Mar 1:26(1):118-123. doi: 10.34763/jmotherandchild.20222601.d-22-00034. Epub 2023 Feb 22 [PubMed PMID: 36803942]
Rodriguez MM. Congenital Anomalies of the Kidney and the Urinary Tract (CAKUT). Fetal and pediatric pathology. 2014 Oct-Dec:33(5-6):293-320. doi: 10.3109/15513815.2014.959678. Epub 2014 Oct 14 [PubMed PMID: 25313840]
Chen CP. Meckel syndrome: genetics, perinatal findings, and differential diagnosis. Taiwanese journal of obstetrics & gynecology. 2007 Mar:46(1):9-14 [PubMed PMID: 17389183]
Verhave JC, Bech AP, Wetzels JF, Nijenhuis T. Hepatocyte Nuclear Factor 1β-Associated Kidney Disease: More than Renal Cysts and Diabetes. Journal of the American Society of Nephrology : JASN. 2016 Feb:27(2):345-53. doi: 10.1681/ASN.2015050544. Epub 2015 Aug 28 [PubMed PMID: 26319241]
Salomon R, Saunier S, Niaudet P. Nephronophthisis. Pediatric nephrology (Berlin, Germany). 2009 Dec:24(12):2333-44. doi: 10.1007/s00467-008-0840-z. Epub 2008 Jul 8 [PubMed PMID: 18607645]
Mikhail MI, Singh AK. Von Hippel-Lindau Syndrome. StatPearls. 2024 Jan:(): [PubMed PMID: 29083737]
Noriega MA, Siddik AB. Trisomy 13. StatPearls. 2024 Jan:(): [PubMed PMID: 32644517]
Zamora EA, Aeddula NR. Tuberous Sclerosis. StatPearls. 2024 Jan:(): [PubMed PMID: 30860727]
Park E, Lee JM, Ahn YH, Kang HG, Ha II, Lee JH, Park YS, Kim NK, Park WY, Cheong HI. Hepatorenal fibrocystic diseases in children. Pediatric nephrology (Berlin, Germany). 2016 Jan:31(1):113-9. doi: 10.1007/s00467-015-3185-4. Epub 2015 Aug 11 [PubMed PMID: 26260382]
Gunay-Aygun M. Liver and kidney disease in ciliopathies. American journal of medical genetics. Part C, Seminars in medical genetics. 2009 Nov 15:151C(4):296-306. doi: 10.1002/ajmg.c.30225. Epub [PubMed PMID: 19876928]