Introduction
The term complement references a set of serum proteins that cooperates with both the innate and the adaptive immune systems to eliminate the bold and tissue pathogens. Like the components of the blood clotting system, complement proteins interact with one another in a catalytic cascade known as complement cascade. The complement system is made up of approximately 40 proteins of an enzymatic, receptor, and regulatory nature, which all participate in a very well-functioning immune system.
At the Institute of Pasteur in the 1890s, Jule Bordet showed that antiserum from sheep causes membrane destruction of bacteria, but this membrane destroying or bacteriolytic activity gets destroyed on heating the antiserum of sheep. The famous immunologist Paul Ehrlich, working independently in Berlin, carried out similar experiments in 1899 and introduced the term “complements” for this heat-labile substance in the sera that were responsible for antimicrobial immunity in addition to antibodies.[1][2]
Issues of Concern
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Issues of Concern
The complement system is a double-edged sword. While it acts as a host defense system, it also has the potential to cause immune and inflammatory diseases due to its inflammatory mediators, which can turn this defensive system into an attacking one for its host. These host-offensive actions become more pronounced with age and become exacerbated by a variety of genetic factors and autoimmune responses. Several complement components are discovered and numbered according to the order of discovery. Various aspects and functions of the complement system and its role in the pathogenesis of autoimmune diseases have been the topic of research. One of the most important reasons for tissue insults and end-organ damage in autoimmune diseases is the excessive activation of the complement pathway.[3]
Paradoxically, deficiencies of specific components of complement pathways also result in manifestations of autoimmune diseases such as systemic lupus erythematosus (SLE).[3] See Figure. Complement Deficiencies Disorder. Advances in sequencing technology and the identification of genetic variants led to the discovery of mutations in components that regulate complement activation in human disease. Striking is the unexpected and remarkable association of mutations in control proteins causing disparate illnesses that predominantly affect the young and the old. An acute endothelial injury syndrome called atypical hemolytic uremic syndrome (aHUS) is a disease often arising in early childhood, while age-related macular degeneration (AMD), which constitutes biological debris in the retina and is a disease of older patients. The complement system plays an etiologic role in the pathogenesis of both disorders.
Cellular Level
Complement components constitute approximately 15% of the globulin protein fraction in the plasma, and their combined concentration can be as high as 3 mg/ml. Also, several regulatory components of the system exist on the cell membranes, so the term complement now embraces glycoproteins distributed among the blood plasma and cell membranes. There are seven functional components of the complement cascade. They are as follows
-
Initiator complement components: As the name suggests, they are the initiators of their respective complement cascades. These proteins do this by attaching to their activating ligands. These ligands are present in a soluble or membrane-bound state. The ligands then get activated to do their respective biological activities by undergoing conformational changes. C1q complex, mannose-binding lectin (MBL), and ficolins are such initiators. See Figure. Biological Effects of Complement Activation.
-
Enzymatic mediators: This component of the complement cascade activates the other members of the complement cascade by their enzymatic activity. They do this either by cleaving or doing conformational changes by binding to these macromolecules. The C3 and C5 convertases are such enzymatic mediators of the complement cascades.
-
Components binding to membrane or opsonins: The enzymatic mediators cleave the complement proteins into two components. One is the larger component named with the suffix “b” and the smaller one with the suffix “a.” Like C3b and C4b are the larger subcomponents of C3 and C4, which act as opsonins. They bind to the microbial cell surface receptors for them and enhance phagocytosis. One exception is for the C2 component.
-
Inflammatory mediators:
The smaller fragment of the complement cascade proteins formed after undergoing activation has a designation with the suffix “a.” Some enhance the local blood supply by binding with endothelial cells of the small blood vessels. They act as inflammatory mediators. They are also known as anaphylatoxins, as their consequences can be destructive in excess.
-
Membrane attack complex:
This protein complex is made up of complement components C5b, C6, C7, and many copies of C9. It is responsible for punching holes into the cell membrane and lysing the microorganism.
-
Complement receptor proteins:
They are present on the cell surfaces and bind to the complement proteins; this leads to signal-specific cell functions.
-
Regulatory complement component:
They are the protective proteins that prevent the complement proteins from overactivity. Examples are factor H and factor I.
Development
In the ensuing years, researchers have discovered that the action of the complement is the result of interactions among a complex group of more than 30 glycoproteins. Most complement components are synthesized in the liver by hepatocytes, although the production of some is also by other cell types, including blood monocytes, tissue macrophages, fibroblasts, epithelial cells of gastrointestinal tracts, and genitourinary tracts.
According to the "evolutionary theory,” the most vital components of nature are revered by multitasking them. The complement system is one such example. C3 and FB, like components of the complement cascade, are the earliest part of the cascade. It evolved half a billion years ago as a primitive form of C3 and has been identified in sponges considered as living fossils.
Organ Systems Involved
There is also a growing appreciation that there is cross-talk between the complement system and other systems, especially the coagulation system.[4] For example, there have been implications of C5a-mediated tissue factor release from neutrophils in the pathogenesis of thrombosis in patients on hemodialysis. With the increase in clinical research, awareness of the role of the complement system, and its role in various diseases, some organ systems showed pronounced susceptibility to complement-mediated injury due to their unique anatomic and functional features that made them conducive to complement activation (see Figure. Schematic of Complement Activations). A broad range of disorders involving various systems has been noted to be known or suspected for complement cascade involvement behind its pathophysiology. They can be acute or chronic, involving a specific tissue or system. Many autoimmune disorders, inflammatory disorders, and age-related disorders are also reported to have complement cascade as its etiopathology. A severe and sudden reaction of complement cascade against PAMPs or DAMPs may lead to hyperinflammation and tissue damage.[5] The too much protective function of complement cascade leads to destructive outcomes like in transplant and biomaterial induced-inflammatory response. This activity affects the outcome of the transplant procedure sometimes by the immediate rejection of the material or, in the long term, by affecting the patient's life by disturbing the function of the foreign material used.
Chronic diseases like atypical hemolytic uremic syndrome (aHUS), Paroxysmal nocturnal hemoglobinuria(PNH), and age-related macular degeneration(AMD) also have complement cascade over activity behind their pathology. Autoimmune diseases like systemic lupus erythematosus (SLE) also have a complement cascade role, although there is a deficiency in clearing the immune complexes, which are supposed to be done by the early complement components of the cascade. This deficiency in clearing the apoptotic cells and debris in the central nervous system leads to the generation of an inflammatory microenvironment that has a role in neurodegenerative disorders like Alzheimer disease.[6] All these malfunctions of the complement cascade of an individual have several causes like alteration in the complement genes and proteins which may be due to mutations that may lead to deletions or deficiencies or may be due to the genetic polymorphisms which may have lead to the gain of functions or loss of functions of the activators and regulators respectively.[7] All such alterations are together known as complotype, which determines the susceptibility of an individual complement cascade involving diseases and disorders.[8]
Function
Complement is a critical part of the host defense machinery that, together with the contact and coagulation systems and the various branches of innate and adaptive immunity, helps to maintain barrier functions and protect against microbial invasion after injury.[9] The role of complement is to detect, tag, and eliminate microbial intruders with almost immediate reactivity but sufficient specificity to avoid damaging host cells.
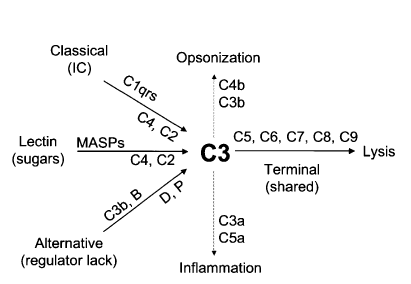
This reactivity and specificity occur via a series of circulating pattern-recognition proteins (PRPs) that sense pathogen-associated molecular patterns (PAMPs) and initiate the complement cascade. The surveillance, immunomodulatory, and effector functions of complement take place through a tightly coordinated network of interactions involving three canonical pathways of activation: the classical pathway, alternative pathway, and lectin pathway. All activation pathways result in the generation of the C3 and C5 convertase enzyme complexes, which cleave C3 into the anaphylatoxin C3a and the opsonin C3b, and C5 into the anaphylatoxin C5a and into C5b, respectively. The deposition of C5b onto a target kicks off membrane attack complex (MAC) formation and target lysis1. The opsonins and anaphylatoxins promote phagocytic uptake of pathogens by scavenger cells and activate neutrophils, monocytes, and mast cells, respectively.[10]
The pro-inflammatory effects of complement are well-acknowledged, but its essential anti-inflammatory properties in immunity are less well appreciated. The anti-inflammatory effects of complement are commonly associated with the lack of complement activation fragment generation, and the recent finding that the absence of anaphylatoxin receptor stimulation during T cell priming induces regulatory T cell development supports this notion.[11] However, new studies suggest that complement has more active, negative regulatory roles in immune responses, in cell and tissue homeostasis, and tissue repair. A picture emerges in which cells provide not only the autocrine activating signals via local complement activation but also simultaneous inhibitory signals. Thus, the generation of initial activation fragments C3a, C5a, and C3b mediates the immune cell-mobilizing effects of complement, but further processed forms of these fragments — for example, inactive C3b (iC3b), designated C3a (C3a-desArg) and C5a-desArg — then engage pathways with negative, tolerogenic and tissue reconstructive capacity.[12] Therefore, it is the balance between these distinct complement signals and the integration of environmental cues (such as growth factor availability and serum complement activation) that instructs cells to keep going or shut down.
Beyond its immune-related functions, complement activity also links to non-immune-related pathways, such as development and tissue homeostasis. During normal development, C3a mediates the mutual cell attraction that is necessary for collective cell migration in Xenopus laevis, and collectin 11 (also known as CLK1) of the lectin complement pathway regulates the migration of neural crest cells.[13] Other studies suggest a role for complement in stem cell or progenitor cell fate decisions. It has been shown that CD46 expression induced by the Notch ligand Delta-like ligand 1 (DLL1) is crucial for human epidermal progenitor cell proliferation and self-renewal and that anaphylatoxins support the maintenance of the pluripotent state of human embryonic stem cells, mediate the mobilization of hematopoietic stem cells from the bone marrow, and promote the migration and proliferation of cardiac pluripotent progenitor cells.[14][15][16] C3a and C5a receptors also facilitate osteoclast differentiation and bone formation.[17] At the level of whole organs, C1q is required for normal neuron maturation as it directs the elimination of excitatory synapses in the brain via ‘synapse pruning’, and it can also protect neurons against fibrillar amyloid beta-mediated injury through the induction of phosphorylated cAMP-response element-binding protein and activator protein 1 (AP-1), which are transcription factors that are associated with neuronal survival and neurite outgrowth.[18][19] By contrast, increased C1q levels correlate with a decline in synaptic plasticity, cognitive function, and tissue regeneration during aging by directly increasing the induction of WNT pathways[20], which suggests that targeting C1q could be a potential approach for the treatment of dementia. The designated form of C3a (C3a-desArg; also known as acylation-stimulating protein) and, as shown more recently, adipocytes and stimulate triglyceride synthesis in adipocytes to produce C3a, and systemic complement has an important homeostatic and regulatory role in metabolic organs such as the pancreas, liver, and adipose tissue. The recently discovered link between intracellular C3aR-mediated signals and the mTOR network suggests that complement may also participate in metabolic sensing. mTOR integrates signals from pathways that sense cellular nutrients, oxygen, and energy levels and translates these signals into appropriate responses, such as apoptosis induction, inhibition of autophagy, and/or cell activation and proliferation. Recent research found that CD46 controls the expression of amino acid and glucose transporters and the assembly of mTOR complex 1 (mTORC1) in activated CD4+ T cells. Furthermore, C3a regulates proteasome activity and, thereby, normal protein turn- over in human retinal epithelial cells88. This relationship suggests that the connection between complement and metabolism may extend to sensing nutrient and/or cellular stress and subsequent modulation of catabolic and anabolic metabolism at the single-cell level.
Mechanism
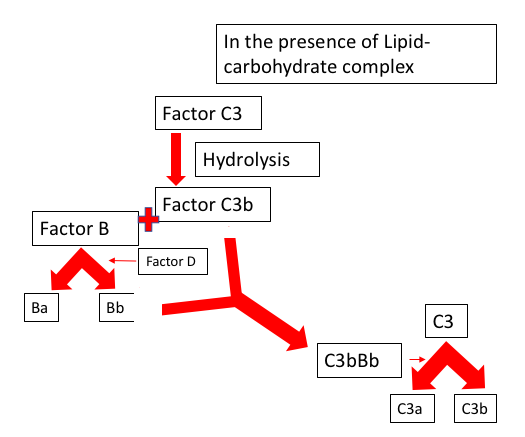
There are three main pathways for complement activation; these are the classical pathway, the lectin pathway, and the alternative pathway. All three pathways are unique in the types of pattern recognizing molecules (PRMs) that initiate a protease cascade leading to the formation of a C3 convertase enzyme complex able to begin the opsonization of complement activating targets. All three pathways are unique in the types of PRMs that initiate a protease cascade leading to the formation of a C3 convertase enzyme complex able to initiate the opsonization of complement activating targets. e.g., C1q, mannose-binding lectin(MBL), ficolins, and properdin are able to recognize pathogen-associated molecular patterns (PAMPs) or DAMPs for autoactivation and initiation of a serine protease cascade, “the complement cascade” leading to opsonization and potentially lysis of the target microbe.[21] See Figure. Opsonization, Complement System.
Initially discovered in 1890, the pathway got its name as “the classical pathway” in the complement cascade. The antigen-antibody complex is the initiation complement component for this pathway. There is an attachment of the C1 to the Fc part of Ig with the help of the C1q subcomponent. This action leads to the commencement of the classical complement cascade leading to many proteolytic cleavage steps. The initial one is the autoactivation of C1r, which cleaves both subcomponents of C1.In the next step, C4 and C2 are cleaved by C1s. The classical pathway C3 convertase gets formed by the combination of newly generated C4b and C2a.C3 convertase then cleaves C3 into C3a and C3b.
Similarly, the C3 convertase of the lectin pathway also gets formed. Here the lectins bind to mannose residues. These mannose-binding lectin-associated serine proteases are substitutes for the C1 enzymatic subcomponents. They target the unique pathogen-related sugar moieties
The capacity of the alternative pathway of self-initiation results from the presence of the internal thioester bond, leading to its deposition on the surface of the pathogen without any prior contact or exposure. This alternative pathway has a self-regulatory mechanism built in it that protects the host cell, and if deposited on the microbial surface, an efficient amplification pathway involving the C3 convertase will create a large amount of C3b to lyse the pathogen.
There are some host regulators of the complement system in both the fluid, i.e., plasma, and the cells and tissues to protect the host cell and tissues from the destructive activity of the complement cascade. Each major step of the complement cascade is under tight control.
The regulatory factors present in the fluid state, i.e., plasma of the host cell, are factor H (FH) and C4b-binding protein (C4BP). Decay-accelerating factor (DAF; CD55),complement receptor 2 (CR2; CD21) complement receptor 1 (CR1; CD35), and membrane cofactor protein (MCP; CD46) are regulatory factors associated with cell membrane
There is a multigene family, the regulators of complement activation (RCAs), which utilize two processes to maintain complement homeostasis. One is called the cofactor activity. In this process, C3b or C4b are permanently cleaved by proteolytic enzymes serine protease factor 1(FI). This inactive those complement components. The second regulatory mechanism is called decay-accelerating activity. Here the catalytic domain (serine protease) of a C3 or C5 convertase is disassociated.
Related Testing
Measurement of the complement system proteins in serum or plasma can divide into four main categories: (a) analysis of complement function or activity; (b) complement factors, individual antigen quantitation; (c) detection of autoantibodies against complement factors; and (d) quantitation of activation fragments, also called split products.[22] Common analytes measured within the classical pathway include CH50 or total complement function, C1q, C2, and C4 individual components (functional and antigen quantitation), and C1-INH (functional and antigen quantitation). Lectin pathway assays are also available, although rarer. Measurement of C3 and the C3 and C4 nephritic factors (autoantibodies against Classical Pathway and Alternative Pathway C3 convertases, respectively) is also of relevance. In the alternative pathway, measurement of AH50 or alternative pathway function, as well as Factor B and Factor H (antigen quantitation) and autoantibodies against Factor B, Factor H, and Factor B split products, Bb and Ba, can be performed. For the analysis of the terminal pathway, C5–C9 individual components quantitation and function and the soluble membrane attack complex (sC5b-9 or sMAC) are also useful. There are several automated and manual methods available that can be used to quantitate the concentration of complement factors. These assays measure the amount of antigen in the sample, commonly reporting the result in milligrams per milliliter or micrograms per deciliter. Detection of autoantibodies to complement factors will aid in differentiating hereditary, genetically mediated complement disorders from acquired conditions. Lastly, the quantification of split products gives an estimation of the activation state of the pathways. It determines if a complement factor has become reduced because of increased consumption or reduced production.
Functional screening tests
Laboratory testing for complement components includes tests for functional activity of the CP (CH50 and its equivalents), the AP (AH50 or APH50), and the MBL pathway. The CH50 has its basis on a hemolytic assay in which an immune complex is formed by adding antibodies that react with a surface antigen on sheep red blood cells. When the antigen-fixed antibodies activate complement on the cell surface, the cell lyses and hemoglobin gets released. Since the formation of the MAC on the cell requires the sequential action of all nine components of the classical (C1, C4, and C2) and terminal (C3, C5, C6, C7, C8, and C9) pathways, titrating the complement source (serum in most cases) so that only a portion of the cells present undergo lysis, the amount of active complement can be calculated, and the results get expressed as the reciprocal of the serum dilution that caused lysis of 50% of the cells in the assay. Two variations of the CH50 are currently in use in clinical laboratories: an assay based on the lysis of liposomes that releases an enzyme that can be read on an analyzer of the sort used for other clinical laboratory tests and solid-phase assays (ELISAs) that detect the final C9 neoantigen that forms when the complete pathway becomes activated. The CH50 is the single best screen for complement abnormalities in that absence or decrease of activity in the CH50 implies that at least one of the necessary components is missing or low. The analogous assay for AP activity, the AH50, is not as widely available as the CH50, but it is useful as a screen for complement deficiency, especially when used in conjunction with the CH50. The AH50 depends on the unique properties of erythrocytes from certain species to provide a surface that promotes activation of the AP, with sequential activation of factors D, B, P, C3, C5, C6, C7, C8, and C9. Properdin is necessary for the stabilization of the C3 convertase (C3bBb), and inefficient activation of the AH50 occurs if P is low or absent. The most common variation of the AH50 uses rabbit red cells in combination with a buffer that blocks the activation of the CP or LP. Like the CH50, the AH50 is a measure of the percentage of rabbit erythrocytes lysed by diluted serum and is expressed in units that represent the dilution that lyses 50% of the cells used in the assay. If both CH50 and AH50 are used to screen for complement deficiency, it minimizes the number of additional tests required to pinpoint the defect. Because both assays include the same 6 terminal components (C3, C5, C6, C7, C8, and C9), the results will be low or absent for both tests if one or more of these components is missing. If a CP component is missing, the CH50 will be low or absent, but the AH50 is normal, whereas if an AP component is low or missing, the reverse will be true. Table II provides a generalized guide to choosing tests for differentiating between acquired and inherited complement deficiencies. Several assays for the C1-Inh function have undergone development. One in common used is an ELISA that measures complexes formed between biotinylated C1s and C1-Inh that get captured on an avidin-coated plate (Quidel). A drawback of this particular assay is that the rare patient might have normal function in the assay but a mutation of the C1-Inh that allows binding of C1s but not of 1 or more of the other enzymes with which the inhibitor reacts. Another test for C1-Inh function relies on the observation that binding of the inhibitor to C1r masks the site on C1r that is recognized by some polyclonal antibodies. After activation of the test serum with aggregated immunoglobulins, the amount of C1r detectable by radial immunodiffusion decreases in proportion to the amount of active C1-Inh present. Functional tests are possible for each of the individual components by using variations on the CH50 or AH50 assays in which an excess of all components except the one under evaluation gets added to the appropriate cells, and the patient’s serum provides the only source of the component in question.
Alternatively, purified components can be added to the patient’s serum to determine which one(s) restores activity. The determination of the function of the MBL pathway is through using an ELISA in which the patient’s serum gets placed into wells coated with mannan. After MBL binds to the mannan-coated surface, the MASP enzymes cleave C4, and the resulting C4b and C4d that get deposited on the plate are measurable by using enzyme-conjugated mAbs.
Quantitative tests for component concentrations
Like most other circulating proteins, the complement components are measurable by standard immunochemical methods available in most laboratories. These include immunoprecipitation assays, including nephelometry, radial immunodiffusion, RIA, and ELISA techniques. The critical points are the specificity of the antibodies used and the reliability of the standard and controls. There are, as yet, very few complement assays that have been standardized and validated for Food and Drug Administration approval. Most laboratories must rely on in-house methods and proven research technology to perform diagnostic procedures for complement analysis. The most definitive method for the evaluation of complement activation is in quantitation of the fragments formed during the enzymatic cleavage steps. Because many of the complement components are acute phase reactants, decreases due to activation might be masked by increases in the synthesis rates during an inflammatory episode. The split products can be used to determine whether activation has occurred because their increase occurs only when the complement enzymes are formed and active. A bonus is that the pathway of activation can be established; C4a and C4d are markers for CP or LP activation, Bb is a marker for AP activation, and C3a, iC3b, C5a, and soluble C5b-9 can be used to determine terminal pathway activation.
Complement autoantibody tests
Testing can determine C1q and C1-Inh autoantibodies by using ELISA methods. The antibodies to C1-Inh bind to the inhibitor molecule and prevent it from attaching to the enzyme, but they don't restrict the enzyme from cleaving the inhibitor. The resulting lower molecular weight inhibitor fragment is detectable by PAGE. There are several assays available for C3v nephritic factors that rely on its function and either measure lysis due to the generation of C3b on a red cell surface or look directly at the amount of C3 that is cleaved when the patient’s serum mixes with normal serum.
Pathophysiology
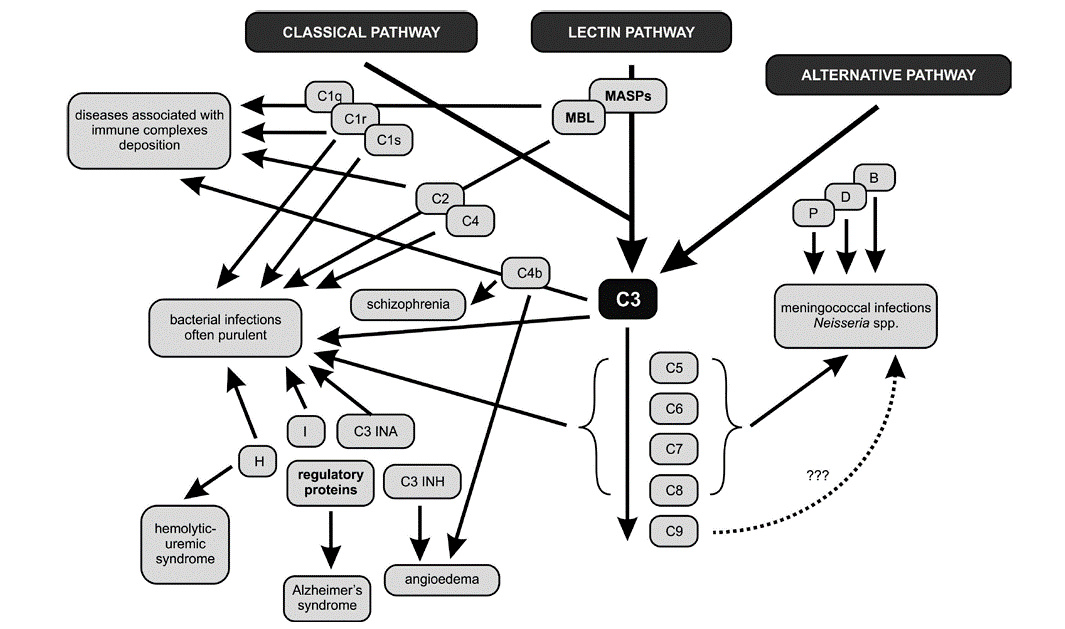
In patients with deficiencies of complement proteins, the greater susceptibility to infer key component of the complement cascade – protein C3 – plays a primary role in the resistance of the host organism to bacterial, viral, fungal, and parasitic infestations. The results of numerous research studies confirm the correlation between C3 deficiency and recurrent infections caused by both Gram-negative (e.g., Neisseria spp., Haemophilus influenza) and Gram-positive (e.g., Streptococcus pneumoniae) bacteria. The particular susceptibility to infections in patients with C3 deficiencies is strictly related to the major role of this component in both the lytic serum properties and opsonophagocytosis. Deficiencies of the late complement proteins involved in the creation of the MAC are related to the occurrence of meningococcal infections. In patients displaying a deficiency of even one of the proteins belonging to the group of C5-C8, recurrent systemic infections, meningitis, purulent otitis media, and bacteremia, usually of severe intensity, may occur.[23] Meningococcal and other bacterial infections also link with properdin deficiency.[24] The absence of the C3INA factor, which controls the activity of C3 in the clinical picture, resembles the state of agammaglobulinemia and promotes the recurrence of bacterial infections. Researchers have also observed that the deficiencies of the alternative pathway’s factors B, D, P, and H lead to increased susceptibility to infections caused by Neisseria spp., Proteus spp., and Pseudomonas spp.
Researchers have studied the roles of complement in killing invading microorganisms for many decades. Since the system can opsonize and lyse foreign particles and cells and generate inflammatory peptides (C3a, C4a, C5a), it has the capacity, if inappropriately activated, to damage host tissues. From the 1970s onwards, its unwanted activities in damaging host tissue were explored in association with diseases such as rheumatoid arthritis, glomerulonephritis, ischemic stroke, myasthenia gravis, multiple sclerosis, and drug-induced lupus. More recently, genetic, genomic, and gene-targeting studies have led to new findings on complement heterozygous deficiencies or common polymorphisms, which lead to subtle alterations in activation and regulation, and are associated with diseases such as age-related macular degeneration (AMD), hemolytic uremic syndrome (HUS) and pre-eclampsia. Knockout studies in mice also highlighted the contributions of complement and related proteins to homeostasis (apoptotic and necrotic cell clearance). Deficiencies in apoptotic cell clearance, which can arise from diminished complement activity, are associated with autoimmune responses, as occur in systemic lupus erythematosus.
AMD affects more than 50 million individuals worldwide and is the leading cause of blindness in the elderly in developed countries.[25] This slowly progressive, degenerative ophthalmological disease of the retina usually manifests after 60 years of age. The prevailing hypothesis is that an overly exuberant inflammatory response, driven by an inadequately regulated complement cascade, significantly contributes to the pathophysiology of AMD. with age-related macular degeneration (AMD). Reduced levels of H and B factors are associated with age-related macular degeneration. Mutations in HTRA1 and CFH genes, encoding the complement regulatory factors H and B, can significantly increase the risk of AMD.[26] Under normal physiological conditions, factor H inhibits the inflammatory response, converting C3b to an inactive iC3b form and also weakens the binding of C3b with factor B. The point mutation in the CFH gene decreases the affinity of its product – factor H to CRP (C-reactive protein), which probably reduces the complement activity regulation in the eye fundus and leads to pathological inflammation in the macula.
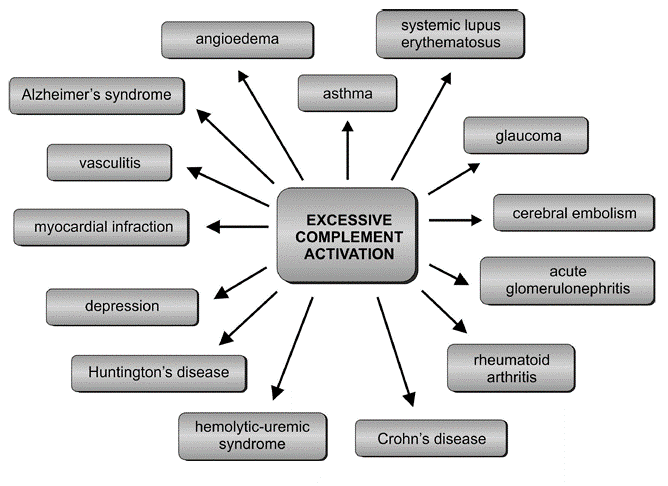
Research has also confirmed the participation of complement components in the atypical form of hemolytic-uremic syndrome (aHUS).[27] aHUS is a disorder caused by excessive complement activation, defects in genes encoding proteins of this system, and the presence of autoantibodies against regulatory proteins that are indispensable for proper complement functioning (see Figure. Excessive Complement Activation).[27] the defects mainly induce ahus in genes primarily encoding factors H and I, deficiencies of which cause severe disorders in the activation of complement resulting in kidney failure. A somewhat less frequent cause of aHUS is mutations in the gene of thrombomodulin, a glycoprotein with anticoagulant properties, playing a role in the inactivation of C3a and C5a anaphylatoxins.[28]
Studies have also observed that the genetically determined deficiency of the C1 inhibitor (C1-INH, C1-esterase inhibitor) occurs in hereditary angioedema (HAE). This is a rare, life-threatening disease of the skin and mucous membranes, manifesting as intermittent, recurrent swelling of the face, genitalia, digestive tract, and larynx. C1-INH is one of the critical regulators of the complement system, coagulation, and kallikrein-kinin cascade.[29] C1-INH inhibits the activity of C1s and C1r, thus preventing the uncontrolled integration of the C2 and C4 components and excessive complement activation through the classical pathway. Its deficiency causes continuous stimulation of this complement route, which is the result of increased self-activation of the C1 protein. In patients with HAE, this activation is persistent and independent of the appearance of clinical symptoms. C1-INH also inhibits active coagulation factor XII and kallikrein resulting in the release of bradykinin, responsible for the formation of edema and pain.[29]
In patients with Alzheimer syndrome, deposition of beta-amyloid plaques in the brain stimulates microglia not only to cytokines secretion, free radicals, and nitric oxide production but also activates the complement system proteins, including the C1 component, Cr3 and Cr4 receptors; this leads to dysfunction of neurons, their degeneration, and, eventually, the patient’s death.[30]
Clinical Significance
Research has uncovered the role of mutation in complement regulatory genes and components in various inflammatory and chronic diseases in the last decade. The advent of next-generation sequencing has helped in these discoveries. But the definitive association of complement cascade is yet to be proved with genetic variants.
The development and approval of eculizumab by the FDA for the treatment of aHUS proves the role of complement cascade involvement in such chronic disorders.
Anti-C5 antibodies have also been found to reduce tissue damage in complement-dependent myocardial infarction and stroke. They are proposed as therapeutic agents in chronic inflammation in, for instance, RA and nephritis.
More research is needed to delineate the complement disease associations further. It will help in the development of more complement-targeted therapeutic agents for diseases like age-related macular disorder.
Media
(Click Image to Enlarge)
Opsonization, Complement System
Contributed by L Thau, MD
(Click Image to Enlarge)
Complement Deficiencies Disorder
Contributed by M Bardhan, MD
(Click Image to Enlarge)
Excessive Complement Activation
Contributed by M Bardhan, MD
(Click Image to Enlarge)
Schematic of Complement Activations
Contributed by M Bardhan, MD
(Click Image to Enlarge)
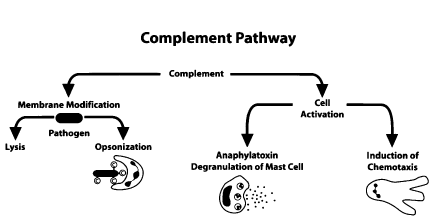
Biological Effects of Complement Activation
Contributed by M Bardhan, MD
References
Kaufmann SH. Immunology's foundation: the 100-year anniversary of the Nobel Prize to Paul Ehrlich and Elie Metchnikoff. Nature immunology. 2008 Jul:9(7):705-12. doi: 10.1038/ni0708-705. Epub [PubMed PMID: 18563076]
Level 3 (low-level) evidenceNesargikar PN, Spiller B, Chavez R. The complement system: history, pathways, cascade and inhibitors. European journal of microbiology & immunology. 2012 Jun:2(2):103-11. doi: 10.1556/EuJMI.2.2012.2.2. Epub 2012 Jun 13 [PubMed PMID: 24672678]
Ballanti E, Perricone C, Greco E, Ballanti M, Di Muzio G, Chimenti MS, Perricone R. Complement and autoimmunity. Immunologic research. 2013 Jul:56(2-3):477-91. doi: 10.1007/s12026-013-8422-y. Epub [PubMed PMID: 23615835]
Level 3 (low-level) evidenceKourtzelis I,Markiewski MM,Doumas M,Rafail S,Kambas K,Mitroulis I,Panagoutsos S,Passadakis P,Vargemezis V,Magotti P,Qu H,Mollnes TE,Ritis K,Lambris JD, Complement anaphylatoxin C5a contributes to hemodialysis-associated thrombosis. Blood. 2010 Jul 29; [PubMed PMID: 20424189]
Ekdahl KN, Soveri I, Hilborn J, Fellström B, Nilsson B. Cardiovascular disease in haemodialysis: role of the intravascular innate immune system. Nature reviews. Nephrology. 2017 May:13(5):285-296. doi: 10.1038/nrneph.2017.17. Epub 2017 Feb 27 [PubMed PMID: 28239169]
Harris CL, Heurich M, Rodriguez de Cordoba S, Morgan BP. The complotype: dictating risk for inflammation and infection. Trends in immunology. 2012 Oct:33(10):513-21. doi: 10.1016/j.it.2012.06.001. Epub 2012 Jun 29 [PubMed PMID: 22749446]
Level 3 (low-level) evidenceBrennan FH, Lee JD, Ruitenberg MJ, Woodruff TM. Therapeutic targeting of complement to modify disease course and improve outcomes in neurological conditions. Seminars in immunology. 2016 Jun:28(3):292-308. doi: 10.1016/j.smim.2016.03.015. Epub 2016 Apr 3 [PubMed PMID: 27049459]
Scott D,Botto M, The paradoxical roles of C1q and C3 in autoimmunity. Immunobiology. 2016 Jun [PubMed PMID: 26001732]
Ricklin D, Lambris JD. Preformed mediators of defense-Gatekeepers enter the spotlight. Immunological reviews. 2016 Nov:274(1):5-8. doi: 10.1111/imr.12497. Epub [PubMed PMID: 27782322]
Sarma JV, Ward PA. The complement system. Cell and tissue research. 2011 Jan:343(1):227-35. doi: 10.1007/s00441-010-1034-0. Epub 2010 Sep 14 [PubMed PMID: 20838815]
Level 3 (low-level) evidenceStrainic MG, Shevach EM, An F, Lin F, Medof ME. Absence of signaling into CD4⁺ cells via C3aR and C5aR enables autoinductive TGF-β1 signaling and induction of Foxp3⁺ regulatory T cells. Nature immunology. 2013 Feb:14(2):162-71. doi: 10.1038/ni.2499. Epub 2012 Dec 23 [PubMed PMID: 23263555]
Level 3 (low-level) evidenceKemper C,Köhl J, Novel roles for complement receptors in T cell regulation and beyond. Molecular immunology. 2013 Dec 15; [PubMed PMID: 23796748]
Level 3 (low-level) evidenceCarmona-Fontaine C, Theveneau E, Tzekou A, Tada M, Woods M, Page KM, Parsons M, Lambris JD, Mayor R. Complement fragment C3a controls mutual cell attraction during collective cell migration. Developmental cell. 2011 Dec 13:21(6):1026-37. doi: 10.1016/j.devcel.2011.10.012. Epub 2011 Nov 24 [PubMed PMID: 22118769]
Level 3 (low-level) evidenceTan DW, Jensen KB, Trotter MW, Connelly JT, Broad S, Watt FM. Single-cell gene expression profiling reveals functional heterogeneity of undifferentiated human epidermal cells. Development (Cambridge, England). 2013 Apr:140(7):1433-44. doi: 10.1242/dev.087551. Epub [PubMed PMID: 23482486]
Borkowska S, Suszynska M, Wysoczynski M, Ratajczak MZ. Mobilization studies in C3-deficient mice unravel the involvement of a novel crosstalk between the coagulation and complement cascades in mobilization of hematopoietic stem/progenitor cells. Leukemia. 2013 Sep:27(9):1928-30. doi: 10.1038/leu.2013.84. Epub 2013 Mar 20 [PubMed PMID: 23511127]
Level 3 (low-level) evidenceLara-Astiaso D,Izarra A,Estrada JC,Albo C,Moscoso I,Samper E,Moncayo J,Solano A,Bernad A,Díez-Juan A, Complement anaphylatoxins C3a and C5a induce a failing regenerative program in cardiac resident cells. Evidence of a role for cardiac resident stem cells other than cardiomyocyte renewal. SpringerPlus. 2012 Dec; [PubMed PMID: 23487597]
Kalbasi Anaraki P, Patecki M, Larmann J, Tkachuk S, Jurk K, Haller H, Theilmeier G, Dumler I. Urokinase receptor mediates osteogenic differentiation of mesenchymal stem cells and vascular calcification via the complement C5a receptor. Stem cells and development. 2014 Feb 15:23(4):352-62. doi: 10.1089/scd.2013.0318. Epub 2013 Dec 11 [PubMed PMID: 24192237]
Level 3 (low-level) evidenceSchafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, Ransohoff RM, Greenberg ME, Barres BA, Stevens B. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron. 2012 May 24:74(4):691-705. doi: 10.1016/j.neuron.2012.03.026. Epub [PubMed PMID: 22632727]
Level 3 (low-level) evidenceBenoit ME, Hernandez MX, Dinh ML, Benavente F, Vasquez O, Tenner AJ. C1q-induced LRP1B and GPR6 proteins expressed early in Alzheimer disease mouse models, are essential for the C1q-mediated protection against amyloid-β neurotoxicity. The Journal of biological chemistry. 2013 Jan 4:288(1):654-65. doi: 10.1074/jbc.M112.400168. Epub 2012 Nov 13 [PubMed PMID: 23150673]
Level 3 (low-level) evidenceNaito AT,Sumida T,Nomura S,Liu ML,Higo T,Nakagawa A,Okada K,Sakai T,Hashimoto A,Hara Y,Shimizu I,Zhu W,Toko H,Katada A,Akazawa H,Oka T,Lee JK,Minamino T,Nagai T,Walsh K,Kikuchi A,Matsumoto M,Botto M,Shiojima I,Komuro I, Complement C1q activates canonical Wnt signaling and promotes aging-related phenotypes. Cell. 2012 Jun 8; [PubMed PMID: 22682250]
Level 3 (low-level) evidenceBajic G, Degn SE, Thiel S, Andersen GR. Complement activation, regulation, and molecular basis for complement-related diseases. The EMBO journal. 2015 Nov 12:34(22):2735-57. doi: 10.15252/embj.201591881. Epub 2015 Oct 21 [PubMed PMID: 26489954]
Cataland SR, Holers VM, Geyer S, Yang S, Wu HM. Biomarkers of terminal complement activation confirm the diagnosis of aHUS and differentiate aHUS from TTP. Blood. 2014 Jun 12:123(24):3733-8. doi: 10.1182/blood-2013-12-547067. Epub 2014 Apr 2 [PubMed PMID: 24695849]
Level 2 (mid-level) evidenceTedesco F. Inherited complement deficiencies and bacterial infections. Vaccine. 2008 Dec 30:26 Suppl 8():I3-8 [PubMed PMID: 19388157]
Stover CM, Luckett JC, Echtenacher B, Dupont A, Figgitt SE, Brown J, Männel DN, Schwaeble WJ. Properdin plays a protective role in polymicrobial septic peritonitis. Journal of immunology (Baltimore, Md. : 1950). 2008 Mar 1:180(5):3313-8 [PubMed PMID: 18292556]
Level 3 (low-level) evidenceWarwick A, Khandhadia S, Ennis S, Lotery A. Age-Related Macular Degeneration: A Disease of Systemic or Local Complement Dysregulation? Journal of clinical medicine. 2014 Nov 3:3(4):1234-57. doi: 10.3390/jcm3041234. Epub 2014 Nov 3 [PubMed PMID: 26237601]
Yang Z, Camp NJ, Sun H, Tong Z, Gibbs D, Cameron DJ, Chen H, Zhao Y, Pearson E, Li X, Chien J, Dewan A, Harmon J, Bernstein PS, Shridhar V, Zabriskie NA, Hoh J, Howes K, Zhang K. A variant of the HTRA1 gene increases susceptibility to age-related macular degeneration. Science (New York, N.Y.). 2006 Nov 10:314(5801):992-3 [PubMed PMID: 17053109]
Level 2 (mid-level) evidenceKavanagh D, Goodship T. Haemolytic uraemic syndrome. Nephron. Clinical practice. 2011:118(1):c37-42. doi: 10.1159/000320901. Epub 2010 Nov 11 [PubMed PMID: 21071971]
Waters AM, Licht C. aHUS caused by complement dysregulation: new therapies on the horizon. Pediatric nephrology (Berlin, Germany). 2011 Jan:26(1):41-57. doi: 10.1007/s00467-010-1556-4. Epub 2010 Jun 18 [PubMed PMID: 20556434]
Banerji A. Current treatment of hereditary angioedema: An update on clinical studies. Allergy and asthma proceedings. 2010 Sep-Oct:31(5):398-406. doi: 10.2500/aap.2010.31.3387. Epub [PubMed PMID: 20929607]
Heneka MT, O'Banion MK, Terwel D, Kummer MP. Neuroinflammatory processes in Alzheimer's disease. Journal of neural transmission (Vienna, Austria : 1996). 2010 Aug:117(8):919-47. doi: 10.1007/s00702-010-0438-z. Epub 2010 Jul 15 [PubMed PMID: 20632195]
Level 3 (low-level) evidence