 Anatomy, Abdomen and Pelvis: Kidney Collecting Ducts
Anatomy, Abdomen and Pelvis: Kidney Collecting Ducts
Introduction
Renal collecting ducts are microscopic tubules that connect to multiple nephrons. Tubular fluid passes through the collecting ducts to reach the calyces and renal pelvis (see Image. Nephron Schematic Illustration). Tubular fluid composition undergoes water and electrolyte reabsorption and secretion. Different cell types line the collecting duct lumen to mediate fluid alterations, regulated by chemical messengers within the kidneys and external organs.
Conditions like diabetes insipidus, hyperaldosteronism, Bartter and Liddle syndromes, and renal tubular acidosis (RTA) involve the renal collecting ducts. Urinary tract obstruction and congenital anomalies are some surgical problems that may impair kidney function that can also involve the collecting ducts. Understanding the anatomy and physiology of the collecting ducts is essential for diagnosing and treating various kidney and electrolyte disorders.
Structure and Function
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Structure and Function
The collecting ducts subdivide into 3 anatomical segments: cortical, outer medullary, and inner medullary. Collecting ducts join distal nephric segments called "collecting tubules." A lobule is comprised of a collecting duct and the group of nephrons it drains. Medullary rays extend into the cortex from the medulla and are composed of loops of Henle and collecting ducts. Collecting ducts drain into the papillary ducts and subsequent minor calyces via papillae at the apices of the pyramids, the area cribrosa. Three cell types predominate within the collecting ducts: the principal and types A and B intercalated cells. Each cell type has characteristically distinct apical and basolateral plasma membrane proteins.[1]
Principal cells contain sodium (epithelial sodium channels or ENaCs) and potassium channels on their apical surfaces. Aldosterone promotes the expression of these ion channels when required to increase sodium reabsorption and potassium secretion. Atrial natriuretic peptide opposes these aldosterone-mediated effects. Principal cells may also express apical aquaporin-2 channels in response to antidiuretic hormone (ADH) stimulation. Aquaporin-2 promotes water reabsorption, increasing tubular fluid concentration (see Image. Normal Urine Production). The principal cells' basolateral surfaces contain sodium-potassium adenosine triphosphatases (Na/K-ATPases), which create intracellular-extracellular electrolyte gradients that promote sodium reabsorption and potassium secretion.[2]
Type A intercalated cells primarily secrete acid into the tubular fluid using apical hydrogen ATPases (H-ATPases) and hydrogen-potassium ATPases (H/K-ATPases). Intracellular carbonic acid dissociates into hydrogen and bicarbonate ions. Bicarbonate enters the circulation via basolateral anion exchanger-1, while hydrogen ions are secreted through the apical H- and H/K-ATPases (see Image. Carbonic Anhydrase 2 Activity in the Type A Renal Intercalated Cell). Secreted hydrogen ions bind to ammonia and hydrogen phosphate to form excretable substances. Aldosterone can potentiate this process by increasing H-ATPase expression.
Type B intercalated cells can be considered the base-secreting equivalents of type A intercalated cells. As in type A intercalated cells, carbonic acid dissociates into hydrogen and bicarbonate ions. Bicarbonate is secreted via an apical anion exchanger called "pendrin." Hydrogen ions enter the circulation via basolateral H-ATPase pumps.[3]
Inner medullary cortical duct cells can express urea channels, which contribute to ADH's overall concentrating effect on tubular fluid. Apical urea transporter-A1 and basolateral urea transporter-A3 expression increase in the presence of this hormone, allowing urea to enter the inner medullary interstitial space and increase urine concentration in this segment. This effect enhances water reabsorption by potentiating passive sodium and chloride reabsorption in the thin ascending limb of the loop of Henle.[4]
Embryology
Nephrogenesis occurs in 3 consecutive phases: pronephros, mesonephros, and metanephros. The mesonephros arises from the intermediate mesoderm by the end of the 4th gestational week. The mesonephros consists of excretory tubules connected to collecting ducts known as mesonephric or Wolffian ducts.[5] Collecting ducts arise from the ureteric bud, which is also the progenitor for the calyces, renal pelvis, ureters, and bladder trigone. The ureteric bud invades the metanephric mesenchyme and undergoes a series of tightly regulated branching events. Branch tips eventually interact with cap mesenchyme cells to promote nephron development and attachment to the collecting system.
Glial cell line-derived neurotrophic factor (GDNF), GDNF's receptor tyrosine kinase RET, fibroblast growth factor, Six1/Sall1 transcription factors, and Wnt signaling pathways are involved in the various stages of ureteric bud growth, invasion, and branching.[6] The mesonephric duct undergoes stimulation through interactions involving the tyrosine kinase inhibitors RET and MET and GDNF and hepatocyte growth factor (HGF), respectively, leading to its evagination and ureteric bud formation. The Wilms tumor-1 (WT1) transcription factor expressed by metanephric mesenchyme helps regulate GDNF and HGF production.[7]
Blood Supply and Lymphatics
The right and left renal arteries diverge from the aorta and divide into 5 segmental arteries: posterior, superior, anterosuperior, anteroinferior, and inferior segmental arteries. These segmental arteries branch into interlobar arteries, which continue to become the arcuate arteries. The collecting ducts span both the cortex and medulla. Thus, more than 1 branch supplies a collecting duct. In the cortex, interlobular arteries bifurcate perpendicularly from the arcuate arteries at cortical and medullary borders. Arcuate arteries give rise to radial arteries. Meanwhile, vasa recta capillaries supply the collecting ducts in the medulla, specifically around the loops of Henle. The renal veins and branches follow the renal arteries' path (see Image. Renal Circulation).[8][9][10]
Lymph from the kidneys primarily passes through the hilar lymphatic route, which involves lymphatic vessels traveling along the renal vasculature toward the aortic and caval nodes. Lymph proceeds to the thoracic duct from these nodes.[11]
Nerves
The kidneys are innervated by autonomic fibers that influence vascular, tubular, and excretory functions. Postganglionic nerves from the renal plexus travel along the renal vasculature to enter the parenchyma. Sympathetic innervation comes from spinal cord levels T11 to L3 and supplies the nephrons and renal vasculature. Sympathetic nerves are concentrated most heavily around the afferent arterioles, thick ascending limbs, and distal convoluted tubules. The vagus nerve contributes parasympathetic fibers.[12][13]
Muscles
Clinically, the musculature and kidneys are connected primarily through creatinine. Creatinine is a muscle breakdown product used clinically to evaluate kidney function.[14] Myoglobin from damaged muscles (eg, in rhabdomyolysis cases) can enter the bloodstream and exert nephrotoxic effects. In contrast, healthy muscles are believed to protect against inflammation-mediated kidney deterioration through several mechanisms. Macroscopically, the kidneys are anterior to the psoas muscles within Gerota’s fascia.[15][16]
Physiologic Variants
Liddle syndrome and carbonic anhydrase II deficiency exhibit collecting tubule variations that affect kidney function. Liddle syndrome is a rare autosomal dominant disorder caused by gain-of-function mutations in genes encoding ENaC subunits. Symptoms include the triad of early-onset hypertension, hypokalemia, and metabolic alkalosis. Normal-to-low aldosterone activity and lack of response to spironolactone distinguish this condition from hyperaldosteronism. The clinician can achieve a definitive diagnosis through genetic testing. Treatment primarily involves maintaining a low-sodium diet and using potassium-sparing diuretics called ENaC blockers.[17]
Carbonic anhydrase II (CAII) mediates carbonic acid formation from water and carbon dioxide in the collecting ducts. Carbonic acid dissociates into hydrogen and bicarbonate, which, as earlier discussed, are handled differently by type A and type B intercalated cells. CAII deficiency is an autosomal recessive disorder that manifests clinically as osteopetrosis, cerebral calcification, and RTA.
RTA may be proximal (type 2), distal (type 1), or combined (type 3). No specific CAII deficiency treatment currently exists, and the condition is typically managed symptomatically.[18][19]
Surgical Considerations
Collecting duct carcinoma (CDC) is an aggressive form of renal cell carcinoma. CDC comprises less than 2% of renal masses and usually carries a poor prognosis. Various gene alterations, including NF2, SETD2, SMARCB1, and CDKN2A, have been identified in CDC cases. Patients with CDC generally undergo cytoreductive nephrectomies, which have been shown to improve survival outcomes, particularly when combined with chemotherapy or radiation).[20][21][22]
Clinical Significance
Besides the aforementioned conditions, many clinical considerations involve the collecting ducts. Some of these conditions are explained below.
Nephrogenic Diabetes Insipidus
Nephrogenic diabetes insipidus (NDI, also called arginine vasopressin resistance) refers to the collecting ducts' inability to concentrate tubular fluid due to an intrinsic renal complication, which may be acquired or genetic. Acquired causes include drug toxicities, electrolyte imbalances, and systemic disorders (eg, autoimmune, amyloidosis, and sarcoidosis).
Lithium is the most common etiology of drug-associated NDI. This substance is believed to downregulate aquaporin-2 expression in principal cells. Genetic causes involve loss-of-function mutations in the genes encoding either vasopressin receptor-2, the ADH receptor in the collecting ducts, or aquaporin-2.
Clinically, NDI manifests as polyuria due to excessive water excretion. The fluid losses lead to secondary polydipsia. The diagnosis may be confirmed with a water deprivation test and a lack of response to desmopressin. Improvement with desmopressin indicates central diabetes insipidus, which is due to impaired ADH release from the posterior pituitary gland. NDI treatment options include addressing reversible secondary causes, thiazide diuretic administration (with or without potassium-sparing diuretics), and low-solute diets.[23]
Syndrome of Inappropriate Antidiuretic Hormone Secretion
SIADH is characterized by excessive fluid retention due to aberrantly high ADH activity. SIADH has many etiologies, including neoplasia, certain drugs, intracranial disorders, and pulmonary disorders. Clinically, SIADH appears as euvolemic hyponatremia with initially increased urine sodium.[24] Solute-free water retention from the collecting duct increases circulating volume, diluting the serum sodium. However, the greater circulating volume prompts a decrease in renin-angiotensin-aldosterone system (RAAS) activity, which restricts sodium and water reabsorption by the principal cells. Thus, the overall effect is the maintenance of normal circulating volume. Sodium excretion eventually matches sodium intake again with homeostasis restoration.[25] Therapeutic strategies mainly involve addressing underlying etiologies and preventing severe hyponatremia. Hyponatremia should not be corrected too rapidly to avoid osmotic demyelination syndrome.[26]
Renal Tubular Acidosis
Types 1, 3, and 4 RTA involve the collecting ducts. Type 1 RTA is classically due to type A intercalated cells' inability to secrete hydrogen ions and reabsorb potassium ions, resulting in hyperchloremic (or normal anion gap) metabolic acidosis and hypokalemia with urine pH greater than 5.3. Type 1 RTA may arise from genetic mutations, systemic diseases (eg, autoimmunity, amyloidosis, and sarcoidosis), and nephrotoxic drug use. Compounds like sodium bicarbonate and potassium citrate can correct acid and potassium imbalances, respectively, but underlying causes should be addressed if present.
Type 4 RTA is a consequence of aldosterone deficiency or resistance. The condition manifests primarily as hyperchloremic metabolic acidosis with hyperkalemia. Type 4 RTA treatment strategies include restricting potassium intake and administering bicarbonate if warranted.
Type 3 RTA is an extremely rare condition with types 1 and 2 RTA characteristics. Type 2 RTA is due to impaired proximal bicarbonate tubular reabsorption. Type 3 RTA can occur as a consequence of acetazolamide misuse or CAII deficiency.[27][28]
Hyperaldosteronism
Aberrantly increased aldosterone activity can result from primary or secondary etiologies. Primary causes include bilateral adrenal hyperplasia and aldosterone-producing adrenal tumors, which produce aldosterone regardless of RAAS activity. Classically, primary hyperaldosteronism manifests as hypervolemia, hypertension, hypokalemia, and metabolic alkalosis. Hypervolemia and hypertension are due to excessive sodium and water retention by principal cells. The resulting alkalosis is due to excessive acid secretion by type A intercalated cells. Hypokalemia is considered a classic hyperaldosteronism symptom, estimated to appear in less than 40% of cases.
Secondary hyperaldosteronism is due to RAAS overactivation. Many etiologies can stimulate RAAS activity, making a definitive symptom profile for secondary hyperaldosteronism difficult to establish. Treatment options include adrenalectomy, mineralocorticoid receptor antagonists, and addressing secondary causes.[29][30][31]
Cushing Syndrome
Cortisol can exert mineralocorticoid effects at sufficiently high levels. In cells containing mineralocorticoid receptors, the enzyme 11β-Hydroxysteroid dehydrogenase type 2 (11β-HSD2) metabolizes cortisol before binding to the receptors. Excess cortisol can overcome cellular 11β-HSD2 capacity, allowing cortisol to bind mineralocorticoid receptors, leading to excessive sodium and water retention in the collecting ducts. This effect contributes to the hypertension seen in Cushing syndrome. Treatment involves addressing the cause of the excess cortisol (eg, by corticosteroid tapering or adrenal tumor resection).[32]
Potassium-Sparing Diuretic Use
Potassium-sparing diuretics interfere with sodium reabsorption at principal cells. The lumen-negative transepithelial potential difference driving subsequent potassium secretion decreases with less sodium reabsorption. Consequently, sodium and water excretion increases, and potassium excretion diminishes.
Potassium-sparing diuretics are classified as mineralocorticoid receptor antagonists or ENaC blockers.[33] Examples of mineralocorticoid receptor antagonists are spironolactone and eplerenone. The intracellular mineralocorticoid receptor is where aldosterone binds to enhance sodium reabsorption and potassium secretion in principal cells. Spironolactone is primarily used for hypertension control, but it may also be used as an adjunct to conventional heart failure regimens. To lessen hyperkalemia risk, a thiazide diuretic may be given concurrenty to lower spironolactone dosage. Spironolactone is highly efficacious but can exert potentially distressing antiandrogenic effects in men like gynecomastia and erectile dysfunction. Eplerenone is understood to have fewer of these antiandrogenic side effects while retaining clinical efficacy.
ENaC blockers are generally less efficacious for treating hypertension than mineralocorticoid receptor antagonists. Thus, ENaC blockers like amiloride and triamterene are usually given with thiazide diuretics. As in mineralocorticoid receptor antagonists, hyperkalemia is a potential side effect. Amiloride can also cause mild gastrointestinal discomfort. Triamterene use can potentially predispose to hyperuricemia and kidney stone formation.[34][35]
Other Issues
Recent research has explored various aspects of collecting duct function. A 2017 study identified the transcription factor TFCP2L1 and its role in collecting duct development. Meanwhile, a 2018 study linked microRNA suppression in collecting duct cells to tubulointerstitial fibrosis. In 2019, research showed tamoxifen's ADH-independent downregulation of aquaporin-2 in the collecting duct. These studies underscore the importance of understanding renal collecting ducts for both scientific and clinical purposes.[36][37][38]
Media
(Click Image to Enlarge)
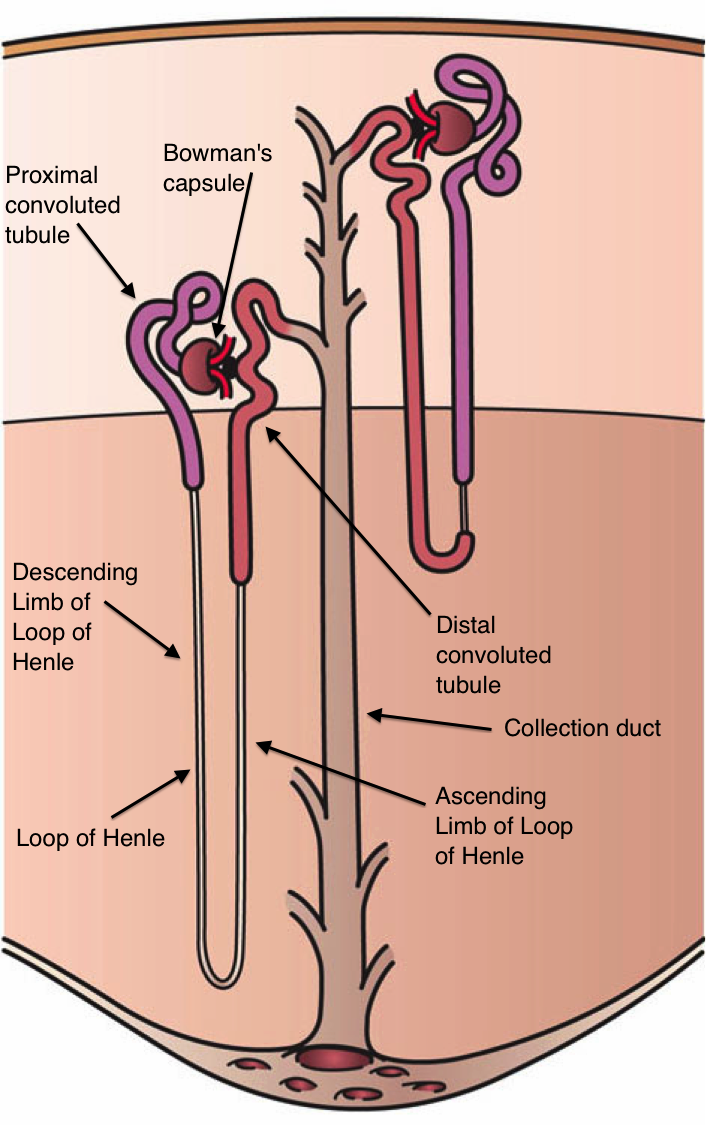
Nephron Schematic Illustration. This image shows the nephron's parts, including the Bowman capsule, proximal convoluted tubule, loop of Henle (with its descending and ascending limbs), distal convoluted tubule, and collecting duct.
Holly Fischer, Public Domain, via Wikimedia Commons
(Click Image to Enlarge)
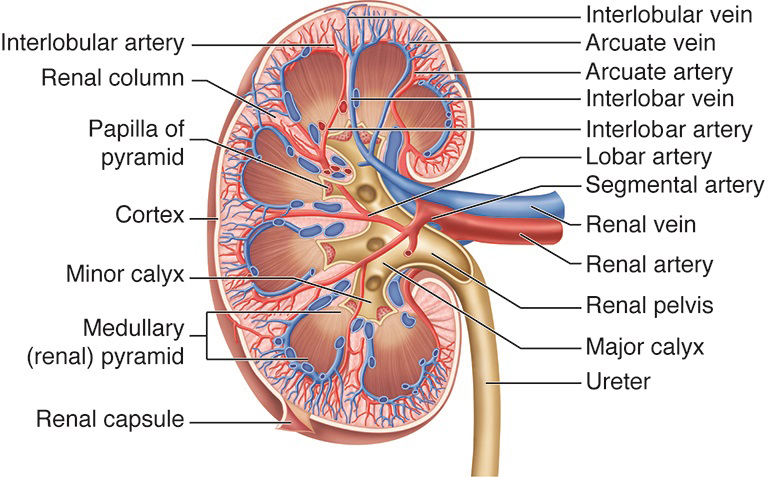
Renal Circulation. This illustration shows the kidney's blood vessels, including the renal, segmental, lobar, interlobar, interlobular, and arcuate arteries and veins. Other structures labeled are the renal columns, pyramidal papilla, cortex, major and minor calyces, renal (medullary) pyramid, renal capsule and pelvis, and ureter.
Cenveo, Public Domain, via AnatomyTOOL.org
(CC BY 4.0 International)
(Click Image to Enlarge)
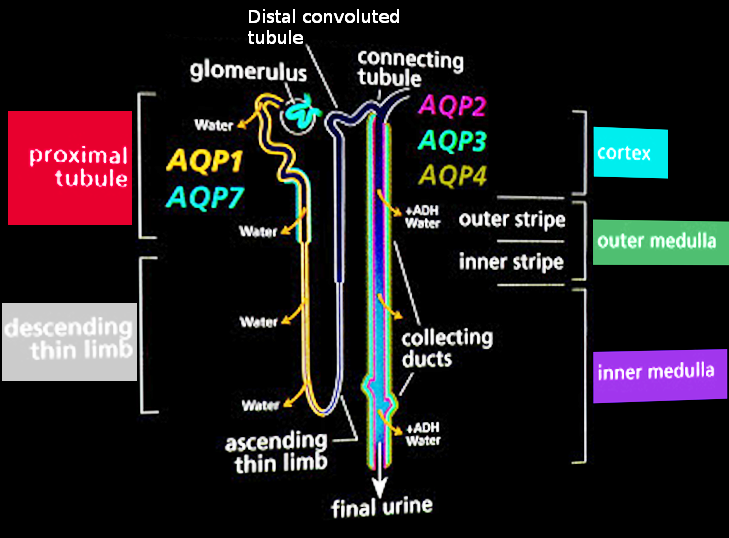
Normal Urine Production. This illustration shows the path of renal filtrate from the glomerulus to the collecting ducts.
Contributed by S Bhimji, MD
(Click Image to Enlarge)
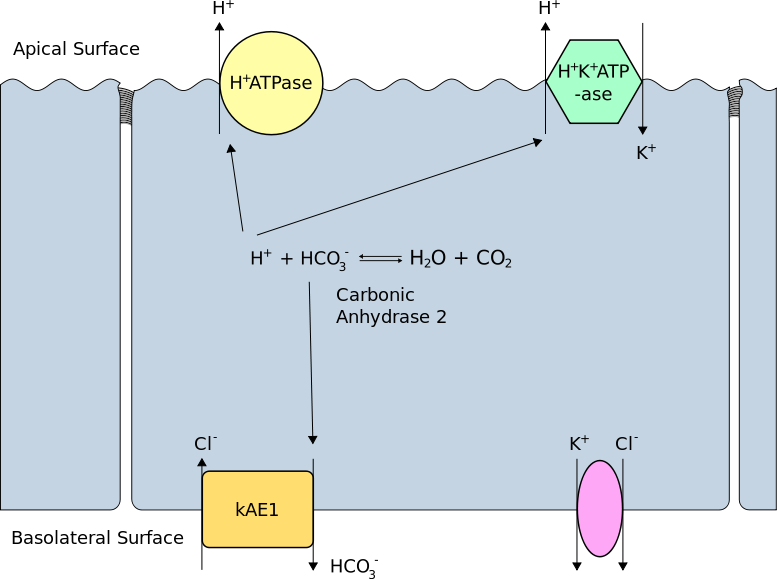
Carbonic Anhydrase 2 Activity in the Type A Renal Intercalated Cell. This illustration shows how carbonic anhydrase forms a proton and a bicarbonate ion from water and carbon dioxide and vice-versa. Protons go to the cell's apical (luminal) surface for secretion via the H-ATPase and H/K-ATPase transporters. Bicarbonate ions enter the interstitium via the basolateral membrane's kidney anion exchangers.
Rswarbrick, Public Domain, via Wikimedia Commons
{kind=link}
References
Staruschenko A. Regulation of transport in the connecting tubule and cortical collecting duct. Comprehensive Physiology. 2012 Apr:2(2):1541-84 [PubMed PMID: 23227301]
Level 3 (low-level) evidencePearce D, Soundararajan R, Trimpert C, Kashlan OB, Deen PM, Kohan DE. Collecting duct principal cell transport processes and their regulation. Clinical journal of the American Society of Nephrology : CJASN. 2015 Jan 7:10(1):135-46. doi: 10.2215/CJN.05760513. Epub 2014 May 29 [PubMed PMID: 24875192]
Level 3 (low-level) evidenceRoy A, Al-bataineh MM, Pastor-Soler NM. Collecting duct intercalated cell function and regulation. Clinical journal of the American Society of Nephrology : CJASN. 2015 Feb 6:10(2):305-24. doi: 10.2215/CJN.08880914. Epub 2015 Jan 28 [PubMed PMID: 25632105]
Level 3 (low-level) evidenceWeiner ID, Mitch WE, Sands JM. Urea and Ammonia Metabolism and the Control of Renal Nitrogen Excretion. Clinical journal of the American Society of Nephrology : CJASN. 2015 Aug 7:10(8):1444-58. doi: 10.2215/CJN.10311013. Epub 2014 Jul 30 [PubMed PMID: 25078422]
Level 3 (low-level) evidenceRehman S, Ahmed D. Embryology, Kidney, Bladder, and Ureter. StatPearls. 2024 Jan:(): [PubMed PMID: 31613527]
Krause M, Rak-Raszewska A, Pietilä I, Quaggin SE, Vainio S. Signaling during Kidney Development. Cells. 2015 Apr 10:4(2):112-32. doi: 10.3390/cells4020112. Epub 2015 Apr 10 [PubMed PMID: 25867084]
Dressler GR. The cellular basis of kidney development. Annual review of cell and developmental biology. 2006:22():509-29 [PubMed PMID: 16822174]
Level 3 (low-level) evidenceApelt K, Bijkerk R, Lebrin F, Rabelink TJ. Imaging the Renal Microcirculation in Cell Therapy. Cells. 2021 May 2:10(5):. doi: 10.3390/cells10051087. Epub 2021 May 2 [PubMed PMID: 34063200]
Jamkar AA, Khan B, Joshi DS. Anatomical study of renal and accessory renal arteries. Saudi journal of kidney diseases and transplantation : an official publication of the Saudi Center for Organ Transplantation, Saudi Arabia. 2017 Mar-Apr:28(2):292-297. doi: 10.4103/1319-2442.202760. Epub [PubMed PMID: 28352010]
Lung K, Lui F. Anatomy, Abdomen and Pelvis: Arteries. StatPearls. 2024 Jan:(): [PubMed PMID: 30247834]
Russell PS, Hong J, Windsor JA, Itkin M, Phillips ARJ. Renal Lymphatics: Anatomy, Physiology, and Clinical Implications. Frontiers in physiology. 2019:10():251. doi: 10.3389/fphys.2019.00251. Epub 2019 Mar 14 [PubMed PMID: 30923503]
Kirkpatrick JJ, Foutz S, Leslie SW. Anatomy, Abdomen and Pelvis: Kidney Nerves. StatPearls. 2024 Jan:(): [PubMed PMID: 29083631]
Soriano RM, Penfold D, Leslie SW. Anatomy, Abdomen and Pelvis: Kidneys. StatPearls. 2024 Jan:(): [PubMed PMID: 29494007]
Shahbaz H, Gupta M. Creatinine Clearance. StatPearls. 2024 Jan:(): [PubMed PMID: 31334948]
Torres PA, Helmstetter JA, Kaye AM, Kaye AD. Rhabdomyolysis: pathogenesis, diagnosis, and treatment. Ochsner journal. 2015 Spring:15(1):58-69 [PubMed PMID: 25829882]
Chitalia V. Muscles Protect the Kidneys. Science translational medicine. 2014 Dec 24:6(268):. pii: 268ec219. doi: 10.1126/scitranslmed.aaa3464. Epub [PubMed PMID: 29977461]
Cui Y, Tong A, Jiang J, Wang F, Li C. Liddle syndrome: clinical and genetic profiles. Journal of clinical hypertension (Greenwich, Conn.). 2017 May:19(5):524-529. doi: 10.1111/jch.12949. Epub 2016 Nov 29 [PubMed PMID: 27896928]
Tinawi M. Pathophysiology, Evaluation, and Management of Metabolic Alkalosis. Cureus. 2021 Jan 21:13(1):e12841. doi: 10.7759/cureus.12841. Epub 2021 Jan 21 [PubMed PMID: 33628696]
Alhuzaim ON, Almohareb OM, Sherbeeni SM. Carbonic Anhydrase II Deficiency in a Saudi Woman. Clinical medicine insights. Case reports. 2015:8():7-10. doi: 10.4137/CCRep.S16897. Epub 2015 Feb 3 [PubMed PMID: 25674028]
Level 3 (low-level) evidenceCiszewski S, Jakimów A, Smolska-Ciszewska B. Collecting (Bellini) duct carcinoma: A clinical study of a rare tumour and review of the literature. Canadian Urological Association journal = Journal de l'Association des urologues du Canada. 2015 Sep-Oct:9(9-10):E589-93. doi: 10.5489/cuaj.2932. Epub 2015 Sep 9 [PubMed PMID: 26425219]
Pal SK, Choueiri TK, Wang K, Khaira D, Karam JA, Van Allen E, Palma NA, Stein MN, Johnson A, Squillace R, Elvin JA, Chmielecki J, Yelensky R, Yakirevich E, Lipson D, Lin DI, Miller VA, Stephens PJ, Ali SM, Ross JS. Characterization of Clinical Cases of Collecting Duct Carcinoma of the Kidney Assessed by Comprehensive Genomic Profiling. European urology. 2016 Sep:70(3):516-21. doi: 10.1016/j.eururo.2015.06.019. Epub 2015 Jul 3 [PubMed PMID: 26149668]
Level 3 (low-level) evidenceSui W, Matulay JT, Robins DJ, James MB, Onyeji IC, RoyChoudhury A, Wenske S, DeCastro GJ. Collecting duct carcinoma of the kidney: Disease characteristics and treatment outcomes from the National Cancer Database. Urologic oncology. 2017 Sep:35(9):540.e13-540.e18. doi: 10.1016/j.urolonc.2017.04.010. Epub 2017 May 8 [PubMed PMID: 28495554]
Kavanagh C, Uy NS. Nephrogenic Diabetes Insipidus. Pediatric clinics of North America. 2019 Feb:66(1):227-234. doi: 10.1016/j.pcl.2018.09.006. Epub [PubMed PMID: 30454745]
Yasir M, Mechanic OJ. Syndrome of Inappropriate Antidiuretic Hormone Secretion. StatPearls. 2024 Jan:(): [PubMed PMID: 29939554]
Zieg J. Pathophysiology of Hyponatremia in Children. Frontiers in pediatrics. 2017:5():213. doi: 10.3389/fped.2017.00213. Epub 2017 Oct 16 [PubMed PMID: 29085814]
Urso C, Brucculeri S, Caimi G. Employment of vasopressin receptor antagonists in management of hyponatraemia and volume overload in some clinical conditions. Journal of clinical pharmacy and therapeutics. 2015 Aug:40(4):376-85. doi: 10.1111/jcpt.12279. Epub 2015 Apr 29 [PubMed PMID: 25924179]
Palmer BF, Kelepouris E, Clegg DJ. Renal Tubular Acidosis and Management Strategies: A Narrative Review. Advances in therapy. 2021 Feb:38(2):949-968. doi: 10.1007/s12325-020-01587-5. Epub 2020 Dec 26 [PubMed PMID: 33367987]
Level 3 (low-level) evidenceAl-Beltagi M, Saeed NK, Bediwy AS, Elbeltagi R, Hasan S, Hamza MB. Renal calcification in children with renal tubular acidosis: What a paediatrician should know. World journal of clinical pediatrics. 2023 Dec 9:12(5):295-309. doi: 10.5409/wjcp.v12.i5.295. Epub 2023 Dec 9 [PubMed PMID: 38178934]
Emmett M. Metabolic Alkalosis: A Brief Pathophysiologic Review. Clinical journal of the American Society of Nephrology : CJASN. 2020 Dec 7:15(12):1848-1856. doi: 10.2215/CJN.16041219. Epub 2020 Jun 25 [PubMed PMID: 32586924]
Vaidya A, Mulatero P, Baudrand R, Adler GK. The Expanding Spectrum of Primary Aldosteronism: Implications for Diagnosis, Pathogenesis, and Treatment. Endocrine reviews. 2018 Dec 1:39(6):1057-1088. doi: 10.1210/er.2018-00139. Epub [PubMed PMID: 30124805]
Cobb A, Aeddula NR. Primary Hyperaldosteronism. StatPearls. 2024 Jan:(): [PubMed PMID: 30969601]
Barbot M, Ceccato F, Scaroni C. The Pathophysiology and Treatment of Hypertension in Patients With Cushing's Syndrome. Frontiers in endocrinology. 2019:10():321. doi: 10.3389/fendo.2019.00321. Epub 2019 May 21 [PubMed PMID: 31164868]
Arumugham VB, Shahin MH. Therapeutic Uses of Diuretic Agents. StatPearls. 2024 Jan:(): [PubMed PMID: 32491770]
Epstein M, Calhoun DA. Aldosterone blockers (mineralocorticoid receptor antagonism) and potassium-sparing diuretics. Journal of clinical hypertension (Greenwich, Conn.). 2011 Sep:13(9):644-8. doi: 10.1111/j.1751-7176.2011.00511.x. Epub 2011 Aug 9 [PubMed PMID: 21896143]
Shah SS, Zhang J, Gwini SM, Young MJ, Fuller PJ, Yang J. Efficacy and safety of mineralocorticoid receptor antagonists for the treatment of low-renin hypertension: a systematic review and meta-analysis. Journal of human hypertension. 2024 Jan 11:():. doi: 10.1038/s41371-023-00891-1. Epub 2024 Jan 11 [PubMed PMID: 38200100]
Level 1 (high-level) evidenceWerth M, Schmidt-Ott KM, Leete T, Qiu A, Hinze C, Viltard M, Paragas N, Shawber CJ, Yu W, Lee P, Chen X, Sarkar A, Mu W, Rittenberg A, Lin CS, Kitajewski J, Al-Awqati Q, Barasch J. Transcription factor TFCP2L1 patterns cells in the mouse kidney collecting ducts. eLife. 2017 Jun 3:6():. pii: e24265. doi: 10.7554/eLife.24265. Epub 2017 Jun 3 [PubMed PMID: 28577314]
Level 2 (mid-level) evidenceHajarnis S, Yheskel M, Williams D, Brefort T, Glaudemans B, Debaix H, Baum M, Devuyst O, Patel V. Suppression of microRNA Activity in Kidney Collecting Ducts Induces Partial Loss of Epithelial Phenotype and Renal Fibrosis. Journal of the American Society of Nephrology : JASN. 2018 Feb:29(2):518-531. doi: 10.1681/ASN.2017030334. Epub 2017 Oct 11 [PubMed PMID: 29021386]
Tingskov SJ, Choi HJ, Holst MR, Hu S, Li C, Wang W, Frøkiær J, Nejsum LN, Kwon TH, Nørregaard R. Vasopressin-Independent Regulation of Aquaporin-2 by Tamoxifen in Kidney Collecting Ducts. Frontiers in physiology. 2019:10():948. doi: 10.3389/fphys.2019.00948. Epub 2019 Aug 9 [PubMed PMID: 31447686]