Introduction
Fuchs endothelial corneal dystrophy (FED) is a bilateral, slowly progressive, often asymmetric corneal disease characterized by deterioration of endothelial cells and development of guttata, which are excrescences of Descemet’s membrane.[1][2] FED slowly progresses to a great extent of endothelial cell loss, subsequent loss of corneal deturgescence, and bilateral corneal edema of the stroma or epithelial layers, leading to ocular pain, glare, halos, and decreased visual acuity.[1][3] FED is the most common form of corneal dystrophy that affects the endothelium. It is also the most common dystrophy that requires keratoplasty for management worldwide.[4][5] In general, the distinguishing feature between “cornea guttata” and FED is the presence of corneal edema in FED.[6] Of note, there is a form of non-guttate form of Fuchs dystrophy in which patients develop corneal edema due to degenerating endothelial cells that do not contain excrescences of Descemet’s membrane. Since non-guttate forms of FED are rare and uncommonly reported in the literature, the majority of the discussion here will focus on FED with the presence of guttata.
FED was first documented in 1910 when Viennese ophthalmologist Ernst Fuchs reported 13 elderly patients with bilateral central clouding.[2] Later, Kraupa described the continuum of corneal changes in FED, and Vogt coined the term “guttata” (Latin: gutta = droplet) in 1921.[7] Since that time, there have been several research studies conducted on guttata and their association with FED. These studies have contributed to the understanding of the pathophysiology of the disease, although more research is necessary to determine its exact mechanism and progression. Additionally, the management of FED has dramatically evolved over the past 20 years and has significantly improved the quality of life of patients.[8] This activity reviews the etiology, epidemiology, pathophysiology, diagnosis, evaluation, management, and prognosis of FED.
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
FED inheritance is in an autosomal dominant manner with variable expressivity and incomplete penetrance.[9][4][10][11] However, almost half of the cases are sporadic. Most patients are unaware of a family history of the disease.[10]
There are several factors that correlate with FED which contribute to its complex pathophysiology. Early-onset FED has heavy influence of genetic factors and is often a familial autosomal dominant disease.[3] Pathogenic mutations in the gene COL8A in the chromosomal position 1p34.3–p32.3 (FCD1) carry associations with the early-onset form of FED pathology, as this mutation affects the structure of Descemet’s membrane. Researchers have identified several genetic mutations in association with late-onset FED; hence, it is more genetically heterogeneous than the early-onset form. Repeat expansion mutations in TCF4, which encodes the E2-2 protein, are associated with FED in many diverse populations; however, they are most prevalent in Caucasians with 80% penetrance. Mutations in the DMPK gene have also been shown to be a FED genetic candidate.
The SLC4A11 gene encodes the ion channel that facilitates water resorption through the endothelium layer and is an important mediator of deturgescence of the cornea. Mutations in this gene can lead to corneal edema and have correlated with FED. Similarly, mutations in the ZEB1 gene, which encodes the transcription factor Zinc finger E-Box binding homeodomain 1, have been associated with late-onset FED and posterior polymorphous corneal dystrophy. The AGBL1 gene encodes the deglutamylase enzyme ATP/GTP binding protein-like 1, and mutations in the gene are associated with FED. Missense mutations in the LOXHD1 gene are associated with progressive hearing loss and corneal endothelial cell dysfunction in FED.[5] Genes KANK4, LAMC1, and ATP1B1 have been linked to FED by their effects on corneal deturgescence.[12] FED can coexist with several conditions, especially with phenotypically similar endothelial corneal dystrophies such as posterior polymorphous corneal dystrophy (PPMD) and congenital hereditary endothelial dystrophy (CHED).[5]
Epidemiology
The incidence and prevalence of FED are challenging to determine, since many patients present with corneal guttata that never progress to FED, and most patients with FED have signs and symptoms that manifest only after several decades. FED initially presents with corneal guttata in the fourth decade of life, but most patients do not require surgical intervention until the sixth or seventh decades.[10] FED is the most common cause of corneal-endothelial transplants worldwide.[13]
Although guttata may occur in up to 4% of patients older than 40 years old in the U.S., they less commonly co-occur with corneal edema, and therefore, many patients with guttata do not receive the diagnosis of FED.[10] There is documented incidence and prevalence of central corneal guttata without edema from several studies. In a 2005 study of adults in Iceland, 11% of women and 7% of men were found to have primary corneal guttata.[14] Another study found that 6.7% of Chinese Singaporeans and 3.7% of Japanese had primary corneal guttata.[15] Lorenzetti et al. found central corneal guttata in 31% of eyes from 20 to 39 years of age, and in 70% of eyes above 40 years of age. Goar et al. found that 9.6% of patients over 40 years of age had guttata. In 1910, Ernst Fuchs speculated that FED likely affected less than one percent of his patients. It is clear that the estimated incidence and prevalence varies greatly, likely due to different clinical definitions of guttata.[2] There are suggestions that FED has a higher prevalence in Europe compared to other areas of the world, and there is a higher proportion of patients that receive transplants secondary to FED in Europe and the USA.[1] Asians show lower rates of FED.[2]
There are several risk factors associated with FED that are not heritable. Age and gender are important factors that influence the development of FED. People older than 40 years old and women have a higher risk of developing FED.[3][16] FED has been reported to have a female to male ratio of 2.5:1 to 3:1.[10] Smoking, exposure to UV light, diabetes are other risk factors that affect the severity of the disease.[5] There is a rare early-onset form of FED that presents in the first decade of life and has a 1 to 1 female to male ratio.[10]
Pathophysiology
The endothelial barrier and pumps are essential in maintaining corneal deturgescence and transparency. Endothelial cell dysfunction plays a significant role in the pathogenesis of FED.
The endothelial layer of the cornea is composed of a monolayer of hexagonal cells that acts as a regulator of water flow, balancing inflow and outflow to prevent corneal swelling. Embryologically, the endothelial cells are derived from neural crest cells and therefore rest in the G1 phase of the cell cycle. The functional endothelium contains around 400 to 500 cells per square millimeter. The aqueous humor provides the cornea with solutes and nutrients, which diffuse through the barrier of endothelial cells via intraocular pressure gradients.[5][10][17]
Tight junctions between endothelial cells limit water diffusion paracellularly; therefore, sodium-potassium ATPase pumps are the primary driver for fluid movement out of the cornea and into the aqueous humor. The sodium-potassium pump is generally considered the endothelial pump; however, it works along with aquaporins, and the SLC4A11 encoded sodium-borate cotransporter. Due to a large number of active transporters, the endothelial cells are extremely metabolically demanding and dependent on ATP, chloride ions, bicarbonate ions, and carbonic anhydrase. For the cornea to remain transparent, the corneal stroma must have less than 3.5 mg of water per mg of dry tissue. Therefore, if the corneal endothelium becomes disrupted, such as if the number of endothelial pumps significantly decreases, the cornea absorbs an uncontrolled amount of water and swells, which results in hazy vision and decreases visual acuity.[5][10][11][10][17]
The pathophysiology of FED involves several proposed mechanisms involving channelopathies, oxidative stress, apoptosis, and the epithelial-mesenchymal transition. Although several proposals for several mechanisms exist, the underlying pathophysiology remains unknown. One of the primary causes of the development of FED is channelopathies in the corneal endothelium, characterized by malfunctions due to mutations in the genes of ion channels, like mutations in SLC4A11, for example.[5] Additionally, endothelial cells in patients with FED have been found to have decreased cytochrome oxidase activity specifically in locations of corneal edema, causing researchers to hypothesize that dysfunction of mitochondria results in an insufficient amount of ATP to drive endothelial pumps.[10] Oxidative stress also appears as an important pathogenetic factor in the development of FED. The cornea has exposure to UV light, which produces reactive oxygen species. The resulting reactive oxygen species cause damage to mitochondrial DNA and nuclear DNA, resulting in apoptosis of endothelial cells.[18][10] Excessive apoptosis evidently plays a role in the degeneration of the endothelium.[11][19][20] The compilation of oxidative DNA damage leads to endothelial cell loss accumulation around the bases of guttata, dysfunction of mitochondria, and ocular tissue degeneration.[5][21]
There is also a possibility that FED is associated with the unfolded protein response (UPR) of the endoplasmic reticulum (ER), in which after unfolded, abnormal proteins accumulate in the ER, a pathway of cellular mechanisms are activated to halt translation, degrading misfolded proteins, and ultimately trigger apoptosis. Few studies have demonstrated that the ER enlarges and several markers of the UPR are overactivated in FED.[22][23] Additionally, the epithelial-mesenchymal transition is thought to be involved in the pathogenesis of FED. Abnormal deposition of extracellular matrix proteins, like collagen and basement membrane, results in a thickening of Descemet’s membrane characteristic of FED.[10] Histopathological analysis of FED reveals endothelial cells that have transformed into the fibroblastic or epithelial phenotype, which can secrete these extracellular matrix proteins.[11] The genes involved in the epithelial-mesenchymal transition include ZEB1 and TCF4.[10][24]
In FED, degenerating endothelial cells produce a pathologic secretion product called hyaline excrescences. These excrescences are called “guttata” and can be seen on Descemet’s membrane, both histologically and clinically.[25] Guttata correlates with membrane thickening. In the early stages, guttata are isolated and generally not confluent, and endothelial cells can compensate for cell loss by undergoing polymegathism.[18][25] Progression occurs over two to three decades. There is an increase in both the number and size of guttata, eventually affecting the peripheral cornea and appearing as confluent guttata.[10] In stage 2, due to loss of endothelium on top of the guttata, these regions of the cornea are not able to maintain deturgescence and corneal thickness increases.[10] As FED progresses, the cornea decompensates, and increased fluid enters, resulting in corneal edema. Corneal swelling is more likely to occur with cell density less than 1000 cells per square mm.[2] In the third stage, epithelial bullae may form due to worsening stromal edema, and these bullae can rupture.[10][25] The final stage includes subepithelial scarring and corneal vascularization.[10][26]
Histopathology
Specular microscopy relies on a smooth, transparent cornea to generate specular reflection; therefore, it is less useful for diseased corneas with significant edema, as in FED.[27] When visible, corneal guttate will appear with dark, oval-to-round areas and may have a small point of reflection in their center.[2] Cell counts will become decreased, and gaps will often remain in between the endothelial cells. In specular microscopy, images are rapidly obtainable, and the procedure is well-established. However, the practice may have limitations due to poor visualization of the corneal stroma in FED patients.
Confocal microscopy provides a superior view of the corneal endothelium compared to specular microscopy. Although it is more useful for evaluation of FED, confocal microscopy not all clinics perform it.[27] This technique uses a gel to reduce light scattering effects at the corneal epithelium; therefore, patients with epithelial disorders or an irregular corneal surface due to significant stromal edema from FED may benefit from this method of examination. On confocal microscopy, “strawberry-like” corneal endothelium and in vivo microstructural changes may be seen. In advanced stages, patients may have fibrosis of Descemet’s membrane, edema of the basal epithelium, and posterior stromal scarring.[28] Anterior stromal cell loss may be visible by histology or confocal microscopy.[26]
Light microscopy will show intracellular epithelial edema, bullous separation, stromal thickening, endothelial loss, and Descemet membrane thickening with posterior nodules, a common finding. Patients with FED only are not expected to have inflammatory cell infiltrates. Electron microscopy will show degenerating keratocytes in the corneal stroma and lipid keratopathy in the majority of cases. Degenerating keratocytes will show vacuolization, dissolution of cytoplasm, loss of intracellular organelles, and nuclear chromatin clumping. The endothelium is attenuated, particularly over the posterior nodules.[18]
Non-guttate forms of Fuchs dystrophy will show many of the same corneal changes as the guttate forms, including endothelial cell pleomorphism and polymegethism, as well as reduced endothelial cell density. However, guttata will be absent. Often, specular microscopy (or confocal microscopy) will be the only way to determine whether a patient has a non-guttate form of FED.[2][29][30] In previous studies, phase contrast microscopy has demonstrated closely-packed excrescences within the thickened lamellae in cases where they are absent on light microscopy, and oxytalan staining has revealed buried guttata on light microscopy. For this reason, some believe that non-guttate and guttate forms of FED are variations of the same disease.[2]
History and Physical
FED usually manifests around the fourth decade of life and can be divided into four stages, which span 20 to 30 years. In the early stages, the majority of patients with FED are asymptomatic,[10] since endothelial cell mechanisms can compensate for cell loss [18]. Routine ophthalmology examination may reveal central corneal guttata as an incidental finding, and biomicroscopy may show guttata, pigment on the posterior corneal surface, and a thickened appearance of Descemet’s membrane [2][10].
Stage 1 is indistinguishable from central corneal guttata (without FED), and it is therefore only identified after the patient progresses to later stages of the disease. In the second stage, the cornea decompensates, and corneal edema occurs. Patients may present with complaints of painless blurry vision and glare that is worse upon waking up in the morning, due to increased corneal hydration that occurs overnight from closed eyelids.[2][10][2][18] Patients may state they use multiple pairs of glasses throughout the day since relative corneal dehydration and therefore hyperopic shift occurs over the day time.[10] Patients may also have a hazy cornea, poor night vision, and pain during blinking. In the third stage, epithelial bullae may be seen on examination, causing further deterioration of vision. These bullae can rupture, causing episodes of pain.[2][10] The final stage includes subepithelial scarring, and patients experience significant vision loss, sometimes as severe as hand motion visual acuity.[2][18] Pain may be absent.[18] This stage is uncommon in the developed world due to the availability of corneal transplants.
Slit-lamp examination is used to assess the extent of guttata, the confluence of guttata (indicating later stages of disease), and the degree of corneal edema, which becomes present at severe stages.[6] Guttata appears centrally in early stages and progress peripherally, more horizontally than vertically, as the disease progresses.[2] With direct illumination, guttata will appear as pinpoint indentations, and with retroillumination, they will appear as small droplets (hence the original term “gutta,” Latin for “droplet”). It is important to distinguish guttata, which are in the central cornea, from Hassall-Henle bodies in the peripheral cornea, which is a normal change that occurs with age. With the progression of the disease, Descemet’s membrane will thicken, and this is best seen with broad tangential illumination. Additionally, guttata will become confluent. Stromal edema will initially appear as gray haze anterior to Descemet’s membrane. As stromal edema increases and pushes the inelastic Descemet’s membrane posteriorly, wrinkles and folds may be present.[2] Full-thickness stromal edema will have a ground-glass appearance.[2]
Slit-lamp examination in FED can also reveal anterior corneal epithelial changes that correlate with disease progression. In stage 2, microcysts will appear on the corneal surface that may be visualized with topical fluorescein and cobalt blue lighting. In stage 3, as extracellular fluid collects beneath the epithelium, epithelial defects may be seen. Subepithelial scarring occurs, and connective tissue pannus forms in stage 4. Over time, the peripheral cornea becomes vascularized, and stromal and epithelial edema decline.[2]
In the few cases of non-guttate FED, guttata will not be present on slit lamp exam; however, marked edema and bullous keratopathy may be apparent.[2][30]
Pachymetry measurement of central corneal thickness (CCT) is often used to aid in decision-making, as a CCT greater than 640 micrometers generally indicates corneal edema. However, patients with physiologically thinner corneas may present with advanced FED and CCT less than 600 micrometers.[6] CCT is useful for monitoring disease progression over time. The corneal central to peripheral thickness (CPTR) ratio may demonstrate a more objective and repeatable metric of FED severity.[6] However, the use of CPTR is not routine in clinical practice.
Evaluation
History, physical examination, pachymetry, and microscopy are key features in evaluating a patient with FED. The first step is to take a thorough history. FED symptoms will typically begin in the sixth to seventh decades of life, and patients will have complaints of blurry vision, glare, and corneal haze. Guttata and corneal edema will be apparent on slit-lamp examination, and later stages may show pathologic changes such as epithelial bullae, scarring, and vascularization.
Specular or confocal microscopy will show endothelial cell loss, and CCT will increase as a result of corneal edema. CCT and endothelial cell count and morphology can be used to monitor disease progression.[10][5] Recently, optical coherence tomography (OCT) has demonstrated utility in documenting guttata in FED. However, quantitative analysis is not possible with current resolution settings, and the image width is restricted to a small region of the central cornea.[1]
Clinicians and patients should monitor FED progression with routine eye exams every six months, and early FED may be managed medically. It is important to note that if patients are scheduled in the morning, their vision will likely be worse compared to afternoon hours. Surgical options may be a consideration for patients with advanced disease associated with decreased function and quality of life.
Patients with FED are typically elderly and therefore may present with several comorbid conditions, including cataracts, age-related macular degeneration, and cardiovascular disease, which can complicate surgical management.[2] The decision to perform cataract surgery alone, or a combined DSEK/phacoemulsification procedure, is dependent on a patient’s CCT and endothelial cell density.
Treatment / Management
There are a number of treatments used to treat FED symptoms of blurry vision and ocular pain. Medical treatments such as hyperosmotic saline drops or ointment can facilitate corneal dehydration. For symptomatic blurry vision in the mornings, some patients find it useful to use a hairdryer to place warm, dry air gently onto the cornea. Other supportive treatments and surgical procedures such as phototherapeutic keratectomy, amniotic membrane transplants, anterior stromal puncture, and conjunctival flaps can be used to relieve painful symptoms, especially those associated with ruptured bullae in later stages of the disease.[1][3](B3)
FED is the primary indication for corneal transplantation in the U.S., and FED accounted for 36% of the nearly 47000 corneal transplants performed in the United States in 2016.[10] Several surgical procedures have shown utility in FED. Penetrating keratoplasty (PK), Descemet’s stripping automated endothelial keratoplasty (DSAEK), and Descemet’s membrane endothelial keratoplasty (DMEK) are the definitive treatments to restore vision.[31] PK was previously used in the management of FED since the 1950s to restore endothelial cells; however, it has several potential complications, such as high postoperative astigmatism, extended visual recovery, suture trauma, persistent epithelial defects, and recurrent refractive surgery.[3][5][8] For these reasons, techniques such as Descemet stripping automated endothelial keratoplasty (DSAEK/DSEK), Descemet membrane endothelial keratoplasty (DMEK), posterior lamellar keratoplasty (PLK), and deep lamellar endothelial keratoplasty (DLEK) have come into favor.[5] DSAEK is currently the most common treatment for endothelial cell dysfunction and results in better visual outcomes than PK as well as minimal changes in astigmatism and spherical equivalent. DMEK involves transplantation of only the endothelial layer and Descemet’s membrane, and it provides the most rapid visual rehabilitation of all of the keratoplasty techniques,[3][5] PLK and DLEK are used less often and are associated with a thick graft-host stromal interface.[32](B3)
More recent potential approaches to managing corneal endothelial cell dysfunction include the use of Rho kinase (ROCK) inhibitor Y-27632, which promotes cell adhesion and proliferation of endothelial cells.[33][34] Corneal collagen cross-linking (CXL) may be used to decrease corneal edema, thereby improving visual acuity and reducing ocular discomfort; however, CXL has had mixed results.[3] Additionally, preliminary research has shown the possible efficacy of lithium, N-acetylcysteine, and sulforaphane in managing FED.[10] Perhaps gene therapies such as adenovirus vector therapy and CRISPR gene editing may be beneficial in managing FED; however, at this time, these treatments pose limited efficacy.[10](B3)
Differential Diagnosis
The differential diagnosis of FED includes [2][3]:
- Primary central corneal guttata
- Aphakic bullous keratopathy (especially with a history of cataract surgery)
- Pseudophakic bullous keratopathy (especially with a history of cataract surgery)
- Recurrent corneal erosions
- Chandler’s syndrome
- Posterior polymorphous dystrophy (PPMD)
- Congenital hereditary endothelial dystrophy (CHED)
- Congenital hereditary stromal dystrophy
- Toxic anterior segment syndrome
- Iridocorneal endothelial (ICE) syndrome
- Hassall-Henle bodies
- Herpetic disciform keratitis
- Pigment dispersion syndrome
- Anterior uveitis
- Interstitial keratitis
- Herpetic stromal keratitis
Staging
Traditionally, staging has its basis on the extent of guttata and presence of corneal edema [26]. Early stages of FED characteristically demonstrate loss of corneal endothelial cells and the formation of guttata of Descemet’s membrane. Later stages of the disease usually involve all layers of the cornea. In 1978, Dr. Jay Krachmer and his team proposed a clinical grading scale for Fuchs Dystrophy to assess the progression of the disease subjectively.
- Grade 0 (G0): Negative; no apparent disease
- Grade 1 (G1): definitive onset of the disease 0-12 central, non-confluent guttata in at least one eye; typically asymptomatic
- Grade 2 (G2): more than 12 central nonconfluent guttata in at least one eye
- Grade 3 (G3): zone of central confluent central guttata 1-2mm in the horizontal plane
- Grade 4 (G4): zone of central confluent central guttata 2-5mm in the horizontal plane
- Grade 5 (G5): zone of central confluent central guttata >5mm with or without edema of the corneal stroma and/or epithelium layer
Additionally, four distinct stages have been defined that span the FED course by two to three decades.[2][3][4]
A description of the four stages appears below:
- Stage 1: asymptomatic; however, slit-lamp examination exhibits central nonconfluent guttata and a thickened Descemet’s membrane
- Stage 2: guttata coalesce; corneal endothelial polymegathism (variation in cell size), pleomorphism (variation in cell shape), and cell loss are present; symptoms of blurry vision and glare, especially when awakening
- Stage 3: dysfunction of the endothelial pump; formation of bullae in epithelial and subepithelial layers; development of corneal edema
- Stage 4: corneal edema results in haziness, scarring, and decreased vision
These staging scales may be losing clinical relevance due to the advent of non-surgical treatments and techniques, allowing for earlier surgical intervention.[26]
Prognosis
FED is a progressive disease, and many patients report significant visual impairment in the sixth or seventh decades of life that requires surgical management. FED is the most common reason for corneal transplant in the United States.[10]
Most FED patients who receive keratoplasty have significant visual improvement.[36][37] A Cochrane review by Nanavaty et al. in 2014 demonstrated no difference in final visual outcome for FED patients who underwent endothelial keratoplasty (i.e., DSEK, DSAEK, DMEK, and femtosecond laser-assisted endothelial keratoplasty) compared to PK.[38] However, it has been demonstrated that patients who undergo endothelial keratoplasty have faster and more reliable visual improvement compared to those who undergo PK,[36][39] as these procedures maintain the structural integrity of the eye. Additionally, several complications can occur with PK. In 2013, DMEK exceeded PK for the treatment of endothelial disorders, and endothelial keratoplasty techniques (DSAEK, DMEK) have since been considered the standard of care for treatment of FED.[40] Patients generally have good visual outcomes.
Complications
There are several complications directly associated with FED, including corneal scarring and decreased visual acuity. Additionally, there are several potential complications of surgical management: graft detachment, rejection, and failure; aqueous leakage; postoperative infection; secondary ocular hypertension from steroid use; cataract formation; wound healing defects; and vitreoretinal complications. PK has significant complications and risks, as patients may develop postoperative astigmatism and wound rupture.[3][5] Due to the complications of PK, endothelial keratoplasty techniques (DSAEK, DMEK) are the standard of care.
Graft detachment rates are reported to range from 0.9% to 36.4% after DSAEK,[41], and a large multicenter found a graft detachment rate of 36.4% after DMEK.[42] Graft detachments are generally treatable with an additional procedure that involves reattaching the graft with air or sulfur hexafluoride. Marques et al. demonstrated that DMEK patients had better-corrected distance visual acuity and a 60% lower rate of rejection compared to DSAEK patients, but DMEK required more rebubblings due to graft detachment.[43] Cataract development may also progress after keratoplasties.[1]
Deterrence and Patient Education
FED is not preventable, and in many cases, the cause is unknown. Patients should return for follow-up appointments to monitor disease progression with routine eye exams. Clinicians should work with their patients to determine when is the appropriate time for medical and surgical intervention. In early stages with corneal decompensation, medical treatment may suffice. Hypertonic saline drops may be used in the mornings to dehydrate the cornea. Some patients find that gently blowing warm air over their corneas with a hairdryer in the mornings is useful to reduce blurry vision. Patients should understand that their disease is progressive, and FED will eventually require surgical intervention.
Enhancing Healthcare Team Outcomes
The management of patients with FED is challenging and complex, and it requires collaboration between ophthalmologists and optometrists on the patient’s care team. FED is a progressive disease, and a patient’s quality of vision and quality of life should be a major consideration when selecting management approaches. Routine eye exams are crucial. Although surgical options are available for advanced disease, there is a possibility of graft failure. In early stages during medical management, the pharmacist can assist with instructing the patients on the use of hyperosmotic eye drops or ointments. If there is a concern with patient compliance, the pharmacist should contact the prescribing as well as provide further patient education. Nursing can also help in this regard, as well as ascertaining and encouraging medication compliance. When the condition progresses to a surgical solution, the ophthalmic surgeon should coordinate with nursing for follow-up care after surgery, so that post-operative care expectations are set and met. Since the FED patient population is typically elderly, patients may benefit from social workers and low vision therapists.
Given the challenge of FED management, it is crucial to employ an interprofessional healthcare team approach, so that collaborative care and information sharing lead to optimal patient outcomes through each stage of the condition. [Level 5]
Media
(Click Image to Enlarge)
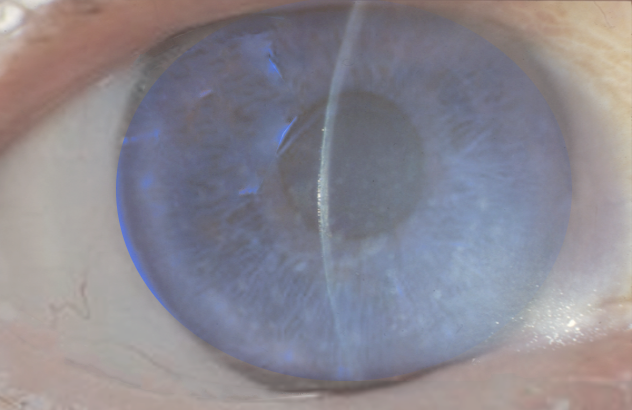
Fuchs endothelial dystrophy Image courtesy Dr Chaigasame
References
Eghrari AO, Gottsch JD. Fuchs' corneal dystrophy. Expert review of ophthalmology. 2010 Apr:5(2):147-159 [PubMed PMID: 20625449]
Adamis AP,Filatov V,Tripathi BJ,Tripathi RC, Fuchs' endothelial dystrophy of the cornea. Survey of ophthalmology. 1993 Sep-Oct; [PubMed PMID: 8235998]
Level 3 (low-level) evidenceFeizi S. Corneal endothelial cell dysfunction: etiologies and management. Therapeutic advances in ophthalmology. 2018 Jan-Dec:10():2515841418815802. doi: 10.1177/2515841418815802. Epub 2018 Dec 7 [PubMed PMID: 30560230]
Level 3 (low-level) evidenceHamill CE,Schmedt T,Jurkunas U, Fuchs endothelial cornea dystrophy: a review of the genetics behind disease development. Seminars in ophthalmology. 2013 Sep-Nov; [PubMed PMID: 24138036]
Nanda GG, Alone DP. REVIEW: Current understanding of the pathogenesis of Fuchs' endothelial corneal dystrophy. Molecular vision. 2019:25():295-310 [PubMed PMID: 31263352]
Level 3 (low-level) evidenceRepp DJ,Hodge DO,Baratz KH,McLaren JW,Patel SV, Fuchs' endothelial corneal dystrophy: subjective grading versus objective grading based on the central-to-peripheral thickness ratio. Ophthalmology. 2013 Apr; [PubMed PMID: 23369486]
Level 2 (mid-level) evidenceSon HS,Villarreal G Jr,Meng H,Eberhart CG,Jun AS, On the origin of 'guttae'. The British journal of ophthalmology. 2014 Sep; [PubMed PMID: 25012533]
Level 3 (low-level) evidenceMoshirfar M, Ding Y, Shah TJ. A Historical Perspective on Treatment of Fuchs' Endothelial Dystrophy: We have Come a Long Way. Journal of ophthalmic & vision research. 2018 Jul-Sep:13(3):339-343. doi: 10.4103/jovr.jovr_94_18. Epub [PubMed PMID: 30090191]
Level 3 (low-level) evidenceZhang J,McGhee CNJ,Patel DV, The Molecular Basis of Fuchs' Endothelial Corneal Dystrophy. Molecular diagnosis [PubMed PMID: 30607871]
Sarnicola C,Farooq AV,Colby K, Fuchs Endothelial Corneal Dystrophy: Update on Pathogenesis and Future Directions. Eye [PubMed PMID: 30005051]
Level 3 (low-level) evidenceZhang J, Patel DV. The pathophysiology of Fuchs' endothelial dystrophy--a review of molecular and cellular insights. Experimental eye research. 2015 Jan:130():97-105. doi: 10.1016/j.exer.2014.10.023. Epub 2014 Nov 1 [PubMed PMID: 25446318]
Afshari NA, Igo RP Jr, Morris NJ, Stambolian D, Sharma S, Pulagam VL, Dunn S, Stamler JF, Truitt BJ, Rimmler J, Kuot A, Croasdale CR, Qin X, Burdon KP, Riazuddin SA, Mills R, Klebe S, Minear MA, Zhao J, Balajonda E, Rosenwasser GO, Baratz KH, Mootha VV, Patel SV, Gregory SG, Bailey-Wilson JE, Price MO, Price FW Jr, Craig JE, Fingert JH, Gottsch JD, Aldave AJ, Klintworth GK, Lass JH, Li YJ, Iyengar SK. Genome-wide association study identifies three novel loci in Fuchs endothelial corneal dystrophy. Nature communications. 2017 Mar 30:8():14898. doi: 10.1038/ncomms14898. Epub 2017 Mar 30 [PubMed PMID: 28358029]
Level 2 (mid-level) evidenceGain P,Jullienne R,He Z,Aldossary M,Acquart S,Cognasse F,Thuret G, Global Survey of Corneal Transplantation and Eye Banking. JAMA ophthalmology. 2016 Feb; [PubMed PMID: 26633035]
Level 3 (low-level) evidenceZoega GM, Fujisawa A, Sasaki H, Kubota A, Sasaki K, Kitagawa K, Jonasson F. Prevalence and risk factors for cornea guttata in the Reykjavik Eye Study. Ophthalmology. 2006 Apr:113(4):565-9 [PubMed PMID: 16581419]
Level 2 (mid-level) evidenceKitagawa K, Kojima M, Sasaki H, Shui YB, Chew SJ, Cheng HM, Ono M, Morikawa Y, Sasaki K. Prevalence of primary cornea guttata and morphology of corneal endothelium in aging Japanese and Singaporean subjects. Ophthalmic research. 2002 May-Jun:34(3):135-8 [PubMed PMID: 12097795]
Zhang X,Igo RP Jr,Fondran J,Mootha VV,Oliva M,Hammersmith K,Sugar A,Lass JH,Iyengar SK, Association of smoking and other risk factors with Fuchs' endothelial corneal dystrophy severity and corneal thickness. Investigative ophthalmology [PubMed PMID: 23882692]
Level 2 (mid-level) evidenceBonanno JA, Molecular mechanisms underlying the corneal endothelial pump. Experimental eye research. 2012 Feb; [PubMed PMID: 21693119]
Level 3 (low-level) evidenceYuen HK, Rassier CE, Jardeleza MS, Green WR, de la Cruz Z, Stark WJ, Gottsch JD. A morphologic study of Fuchs dystrophy and bullous keratopathy. Cornea. 2005 Apr:24(3):319-27 [PubMed PMID: 15778606]
Borderie VM, Baudrimont M, Vallée A, Ereau TL, Gray F, Laroche L. Corneal endothelial cell apoptosis in patients with Fuchs' dystrophy. Investigative ophthalmology & visual science. 2000 Aug:41(9):2501-5 [PubMed PMID: 10937560]
Li QJ, Ashraf MF, Shen DF, Green WR, Stark WJ, Chan CC, O'Brien TP. The role of apoptosis in the pathogenesis of Fuchs endothelial dystrophy of the cornea. Archives of ophthalmology (Chicago, Ill. : 1960). 2001 Nov:119(11):1597-604 [PubMed PMID: 11709009]
Halilovic A, Schmedt T, Benischke AS, Hamill C, Chen Y, Santos JH, Jurkunas UV. Menadione-Induced DNA Damage Leads to Mitochondrial Dysfunction and Fragmentation During Rosette Formation in Fuchs Endothelial Corneal Dystrophy. Antioxidants & redox signaling. 2016 Jun 20:24(18):1072-83. doi: 10.1089/ars.2015.6532. Epub 2016 Apr 11 [PubMed PMID: 26935406]
Jun AS,Meng H,Ramanan N,Matthaei M,Chakravarti S,Bonshek R,Black GC,Grebe R,Kimos M, An alpha 2 collagen VIII transgenic knock-in mouse model of Fuchs endothelial corneal dystrophy shows early endothelial cell unfolded protein response and apoptosis. Human molecular genetics. 2012 Jan 15; [PubMed PMID: 22002996]
Level 3 (low-level) evidenceKim EC, Toyono T, Berlinicke CA, Zack DJ, Jurkunas U, Usui T, Jun AS. Screening and Characterization of Drugs That Protect Corneal Endothelial Cells Against Unfolded Protein Response and Oxidative Stress. Investigative ophthalmology & visual science. 2017 Feb 1:58(2):892-900. doi: 10.1167/iovs.16-20147. Epub [PubMed PMID: 28159976]
Okumura N,Minamiyama R,Ho LT,Kay EP,Kawasaki S,Tourtas T,Schlötzer-Schrehardt U,Kruse FE,Young RD,Quantock AJ,Kinoshita S,Koizumi N, Involvement of ZEB1 and Snail1 in excessive production of extracellular matrix in Fuchs endothelial corneal dystrophy. Laboratory investigation; a journal of technical methods and pathology. 2015 Nov; [PubMed PMID: 26302187]
WOLTER JR, LARSON BF. Pathology of cornea guttata. American journal of ophthalmology. 1959 Jul:48(1, Part 2):161-9 [PubMed PMID: 13670277]
Amin SR, Baratz KH, McLaren JW, Patel SV. Corneal abnormalities early in the course of Fuchs' endothelial dystrophy. Ophthalmology. 2014 Dec:121(12):2325-33. doi: 10.1016/j.ophtha.2014.07.001. Epub 2014 Aug 22 [PubMed PMID: 25156138]
Level 2 (mid-level) evidenceHara M,Morishige N,Chikama T,Nishida T, Comparison of confocal biomicroscopy and noncontact specular microscopy for evaluation of the corneal endothelium. Cornea. 2003 Aug; [PubMed PMID: 12883342]
Level 2 (mid-level) evidenceGrupcheva CN, Craig JP, Sherwin T, McGhee CN. Differential diagnosis of corneal oedema assisted by in vivo confocal microscopy. Clinical & experimental ophthalmology. 2001 Jun:29(3):133-7 [PubMed PMID: 11446452]
Level 3 (low-level) evidenceAbbott RL, Fine BS, Webster RG Jr, Paglen PG, Spencer WH. Specular microscopic and histologic observations in nonguttate corneal endothelial degeneration. Ophthalmology. 1981 Aug:88(8):788-800 [PubMed PMID: 6976545]
Level 3 (low-level) evidenceBrooks AM, Grant G, Gillies WE. A comparison of corneal endothelial morphology in cornea guttata, Fuchs' dystrophy and bullous keratopathy. Australian and New Zealand journal of ophthalmology. 1988 May:16(2):93-100 [PubMed PMID: 3263136]
Vedana G, Villarreal G Jr, Jun AS. Fuchs endothelial corneal dystrophy: current perspectives. Clinical ophthalmology (Auckland, N.Z.). 2016:10():321-30. doi: 10.2147/OPTH.S83467. Epub 2016 Feb 18 [PubMed PMID: 26937169]
Level 3 (low-level) evidenceFernandez MM, Afshari NA. Endothelial Keratoplasty: From DLEK to DMEK. Middle East African journal of ophthalmology. 2010 Jan:17(1):5-8. doi: 10.4103/0974-9233.61210. Epub [PubMed PMID: 20543930]
Macsai MS, Shiloach M. Use of Topical Rho Kinase Inhibitors in the Treatment of Fuchs Dystrophy After Descemet Stripping Only. Cornea. 2019 May:38(5):529-534. doi: 10.1097/ICO.0000000000001883. Epub [PubMed PMID: 30720541]
Koizumi N, Okumura N, Ueno M, Kinoshita S. New therapeutic modality for corneal endothelial disease using Rho-associated kinase inhibitor eye drops. Cornea. 2014 Nov:33 Suppl 11():S25-31. doi: 10.1097/ICO.0000000000000240. Epub [PubMed PMID: 25289721]
Level 3 (low-level) evidenceKrachmer JH, Purcell JJ Jr, Young CW, Bucher KD. Corneal endothelial dystrophy. A study of 64 families. Archives of ophthalmology (Chicago, Ill. : 1960). 1978 Nov:96(11):2036-9 [PubMed PMID: 309758]
Trousdale ER, Hodge DO, Baratz KH, Maguire LJ, Bourne WM, Patel SV. Vision-related quality of life before and after keratoplasty for Fuchs' endothelial dystrophy. Ophthalmology. 2014 Nov:121(11):2147-52. doi: 10.1016/j.ophtha.2014.04.046. Epub 2014 Jul 9 [PubMed PMID: 25015214]
Level 2 (mid-level) evidencevan der Meulen IJ, Patel SV, Lapid-Gortzak R, Nieuwendaal CP, McLaren JW, van den Berg TJ. Quality of vision in patients with fuchs endothelial dystrophy and after descemet stripping endothelial keratoplasty. Archives of ophthalmology (Chicago, Ill. : 1960). 2011 Dec:129(12):1537-42. doi: 10.1001/archophthalmol.2011.247. Epub 2011 Aug 8 [PubMed PMID: 21825178]
Level 2 (mid-level) evidenceNanavaty MA, Wang X, Shortt AJ. Endothelial keratoplasty versus penetrating keratoplasty for Fuchs endothelial dystrophy. The Cochrane database of systematic reviews. 2014 Feb 14:2(2):CD008420. doi: 10.1002/14651858.CD008420.pub3. Epub 2014 Feb 14 [PubMed PMID: 24526345]
Level 1 (high-level) evidencePrice MO, Gupta P, Lass J, Price FW Jr. EK (DLEK, DSEK, DMEK): New Frontier in Cornea Surgery. Annual review of vision science. 2017 Sep 15:3():69-90. doi: 10.1146/annurev-vision-102016-061400. Epub 2017 Jul 11 [PubMed PMID: 28697678]
Röck T, Landenberger J, Bramkamp M, Bartz-Schmidt KU, Röck D. The Evolution of Corneal Transplantation. Annals of transplantation. 2017 Dec 15:22():749-754 [PubMed PMID: 29242495]
Nahum Y, Leon P, Mimouni M, Busin M. Factors Associated With Graft Detachment After Primary Descemet Stripping Automated Endothelial Keratoplasty. Cornea. 2017 Mar:36(3):265-268. doi: 10.1097/ICO.0000000000001123. Epub [PubMed PMID: 28079683]
Ang M, Wilkins MR, Mehta JS, Tan D. Descemet membrane endothelial keratoplasty. The British journal of ophthalmology. 2016 Jan:100(1):15-21. doi: 10.1136/bjophthalmol-2015-306837. Epub 2015 May 19 [PubMed PMID: 25990654]
Marques RE, Guerra PS, Sousa DC, Gonçalves AI, Quintas AM, Rodrigues W. DMEK versus DSAEK for Fuchs' endothelial dystrophy: A meta-analysis. European journal of ophthalmology. 2019 Jan:29(1):15-22. doi: 10.1177/1120672118757431. Epub 2018 Apr 16 [PubMed PMID: 29661044]
Level 1 (high-level) evidence