Introduction
Glycogen storage disease type II (GSD2, Pompe Disease) is a recessive metabolic disorder, creating glycogen deposits inside lysosomes within the muscular tissue[1].
This disease is either classified as early (infantile, classic) or late-onset (non-classic). Early-onset has a severe presentation and is likely to feature a fatal outcome, should prompt treatment not be available. A common cause of lethality for both onsets is respiratory insufficiency, which manifests at different ages in late-onset. Another important cause of lethality in infantile-onset is left ventricular outflow obstruction.
Preventive treatment consists of enzyme replacement therapy (alglucosidase alfa)[2], maintaining an updated immunization schedule, and respiratory syncytial virus (RSV) prophylaxis. Adults developing signs of respiratory insufficiency can benefit from respiratory exercises or assisted mechanical ventilation, which may also be convenient for children.
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
The GAA gene on chromosome 17q25.3 synthesizes acid alfa-glucosidase, which is a lysosomal enzyme catalyzing alpha 1,4 and alpha 1,6 linkages of lysosomal glycogen. Mutations in such a gene inherited in an autosomal recessive fashion, lead to unstable mRNA translating into a deficient or null product.
Epidemiology
Incidence varies according to ethnicity and region; it is estimated to be 1:40,000 for the U.S.[1], with 1:14,000 for African Americans. The numbers mentioned above include all GSD2 forms.
Certain populations could also have a particular mutation associated with their ethnic group. For example, p.Asp654Glu is mainly present in Taiwanese and Chinese patients, while p.Arg854Ter is present in individuals of African descent. [3].
Pathophysiology
Although a precise mechanism has not been described, lysosomal glycogen accumulation leads to impairment of striated muscular cells[1]. Initially, affected areas involve active voluntary muscles (extremities), progressing to cardiac muscle, and lastly, the diaphragm.
History and Physical
Two phenotypes have been described, early (infantile, classic) and late (non-classic) onset[3].
Early-onset is characterized by exhibiting symptoms before the age of 1 year[1]. Common problems are hypotonia/muscle weakness with subsequent motor delay (96%), cardiomegaly (92%, commonly hypertrophic cardiomyopathy)[1], hepatomegaly (90%), macroglossia (62%), poor feeding/failure to thrive (53% to 57%); respiratory infections or dyspnea lead to respiratory failure, which is the most common cause of death in this disease[4]. It has also been associated with hearing loss.
Late-onset patients commonly exhibit symptoms in childhood or beyond in a variable manner, though usually as proximal muscle weakness. Symptom progression is slower but ultimately leads to lower limb weakness and respiratory failure due to diaphragm affection[5]. It is not common to find cardiac involvement. However, a small group of adults has presented with signs of arterial problems. A clue for diagnosing this illness in adolescents or adults would be a history of "clumsiness" while performing physical activities.
Evaluation
For early-onset in areas where newborn screening is not available (see below), creatine kinase (CK) serum and urinary oligosaccharides can be the first step[4].
An acid alpha-glucosidase enzyme assay can be performed in whole or dried blood[6][7]. However, the standard gold test is enzyme analysis from cultured fibroblasts or muscle[5][7], which could have an important setback, since it requires a longer time for result delivery. It is estimated that residual levels below 1% of activity are linked to infant onset, while residual levels above 2% to 40% are linked with late-onset. However, discrimination must be taken into account, since cultured fibroblast methodology is more accurate of the two.
Acid alpha-glucosidase (GAA) gene sequencing confirms the diagnosis[4][7][4]. A thoughtful process for ordering the test should be taken into account since there are populations (see above) who would benefit from targeted gene sequence analysis rather than complete sequence analysis with deletion/duplication.
Due to the variant and often misleading nature of the late-onset phenotype, mimicking or sometimes overlapping with other muscular disorders, initial testing most of the time includes EMG, nerved conduction studies (results are normal for motor and sensory pathways); and muscle biopsy[7] with PAS staining in search for lysosomal glycogen accumulation.
Treatment / Management
Following diagnosis, the initial workup should consist of chest radiography, electrocardiography, an echocardiogram to evaluate cardiac status in the early onset type.
The reduction of vital capacity can be assessed in late-onset, which is a difficult approach with infants since the procedure requires instruction compliance. For all onsets, a prompt response should be provided to patients who develop a cough, wheezing, or shortness of breath[3].(B3)
For feeding difficulties consider an orogastric or nasogastric tube, as well as video swallow studies (late-onset) to prevent aspiration and/or gastroesophageal reflux.
Enzyme replacement therapy (ERT) is currently the most reliable treatment. By providing an analogous enzyme, lysosomal glycogen accumulation in cardiac and skeletal muscle is actively reduced, though diagnosis timing is the main determinant factor for treatment response. CRIM (cross-reactive immunologic material) status is to be determined either prior to or during the first administrations of ERT. CRIM testing is important since patients who develop antibodies against the provided enzyme can have a reduced effect. Different protocols have been created to reduce this response, which can consist of antihistamines, steroids, and other immunomodulators. Dosing for ERT in its commercial forms, ranging from 20 to 40 mg/kg/dose every two weeks[2].(A1)
Treatment of the different complications should be tailored particularly to each case. Cardiac affected patients should avoid digoxin or inotropes initially since they can worsen left ventricular outflow status. Muscle weakness is greatly benefited from physical therapy, mainly to avoid contractures of the pelvic girdle, which can require surgical management. Speech and swallow therapy, CPAP, and BiPAP are often required in patients with the late-onset phenotype.
Differential Diagnosis
The differential diagnosis of early-onset includes (differences are written in parenthesis): spinal muscular atrophy 1 (no cardiac involvement), Danon disease (X-linked inherited)[3].
For late-onset: limb-girdle muscular dystrophy (no affection of axial muscles), Duchenne-Becker muscular dystrophy (X-linked inherited). Other glycogen storage disorders can be considered; however, the main difference consists of a lack of hypoglycemia in GSD2.
Pearls and Other Issues
Newborn Screening
Few countries have adopted this preventive method. Within the United States, it is not universally performed, but steps are being taken to do so. The technique consists of enzyme analysis on dried blood spots through mass spectrometry. Reasoning to perform it universally is that, while diagnosed early, it can improve the prognosis dramatically. This is particularly true in early-onset where cardiac and motor development present remarkable positive results when ERT is administered within the first two weeks of life. Respiratory support is not as needed with patients who receive ERT prior to six months of age. Nonetheless, patients with particular muscle fiber resistance to therapy, antibodies against ERT, or CRIM negativity will prove to be on the poor prognostic side.
Patients with late-onset have a better prognosis overall, with their main factors being muscle weakness and respiratory insufficiency. On ERT, patients showed an optimal response along with increased quality of life.
In both groups, no long-term studies have been performed, given the relative novelty of ERT.
Patients who will be in stressful situations, like pregnancy or surgery, should be monitored closely with cardiac and respiratory surveillance.
Novel therapies are underway, with adenovirus-based gene therapy being one of the most recent ones to improve the respiratory status and reduce the need for ventilatory assistance[8].
Enhancing Healthcare Team Outcomes
Glycogen storage diseases are very rare. All members of the interprofessional team including nurses and clinicians need to be aware of the signs and symptoms and report their findings to the team. Team members may include cardiologists, pulmonologists, physiatrists, neurologists, and physical therapists. Pharmacists should be involved in enzyme replacement therapy (ERT). Early diagnosis and treatment will result in the best outcome. [Level 5]
Media
(Click Image to Enlarge)
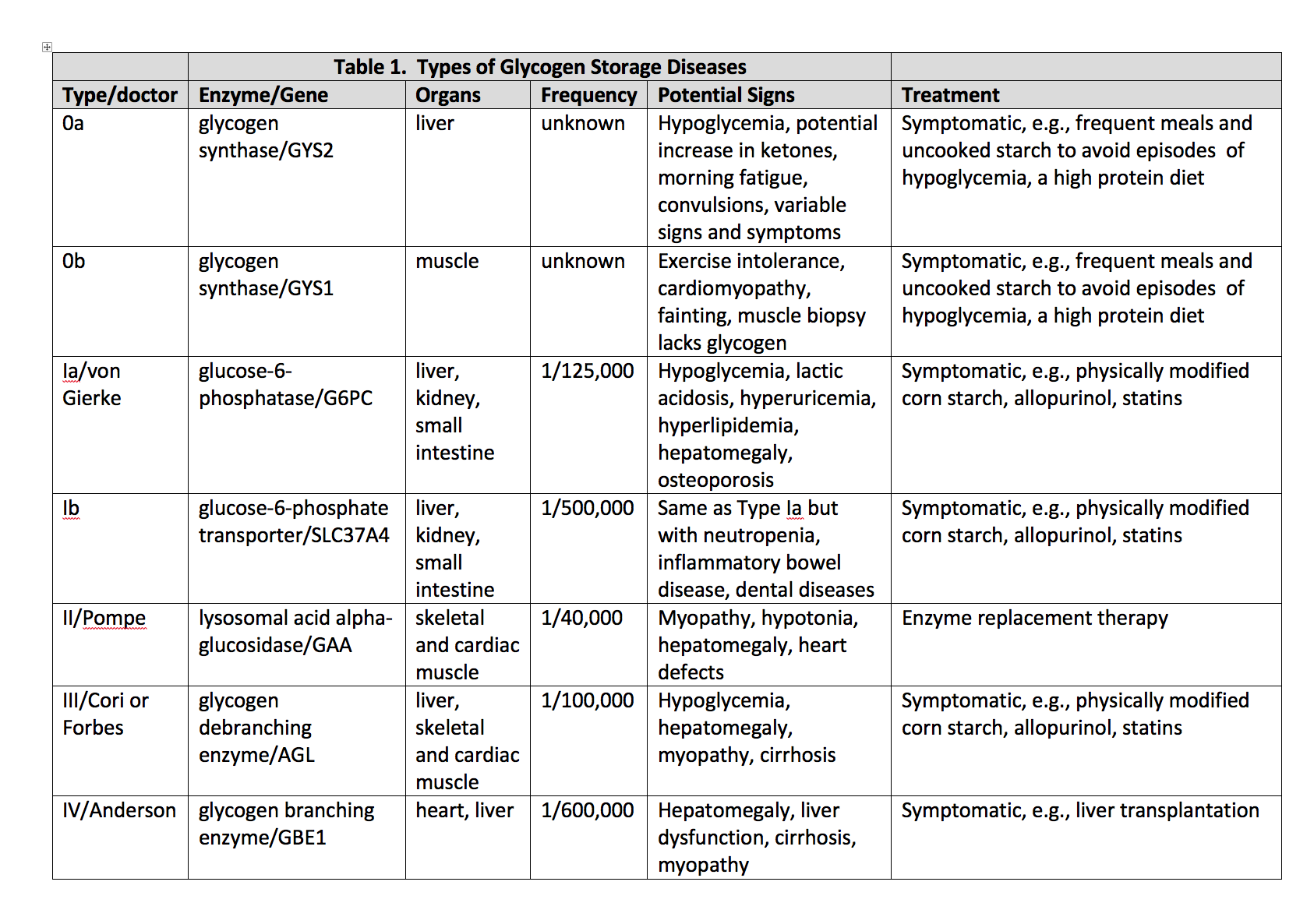
Types of glycogen storage diseases Contributed by William L. Stone
(Click Image to Enlarge)
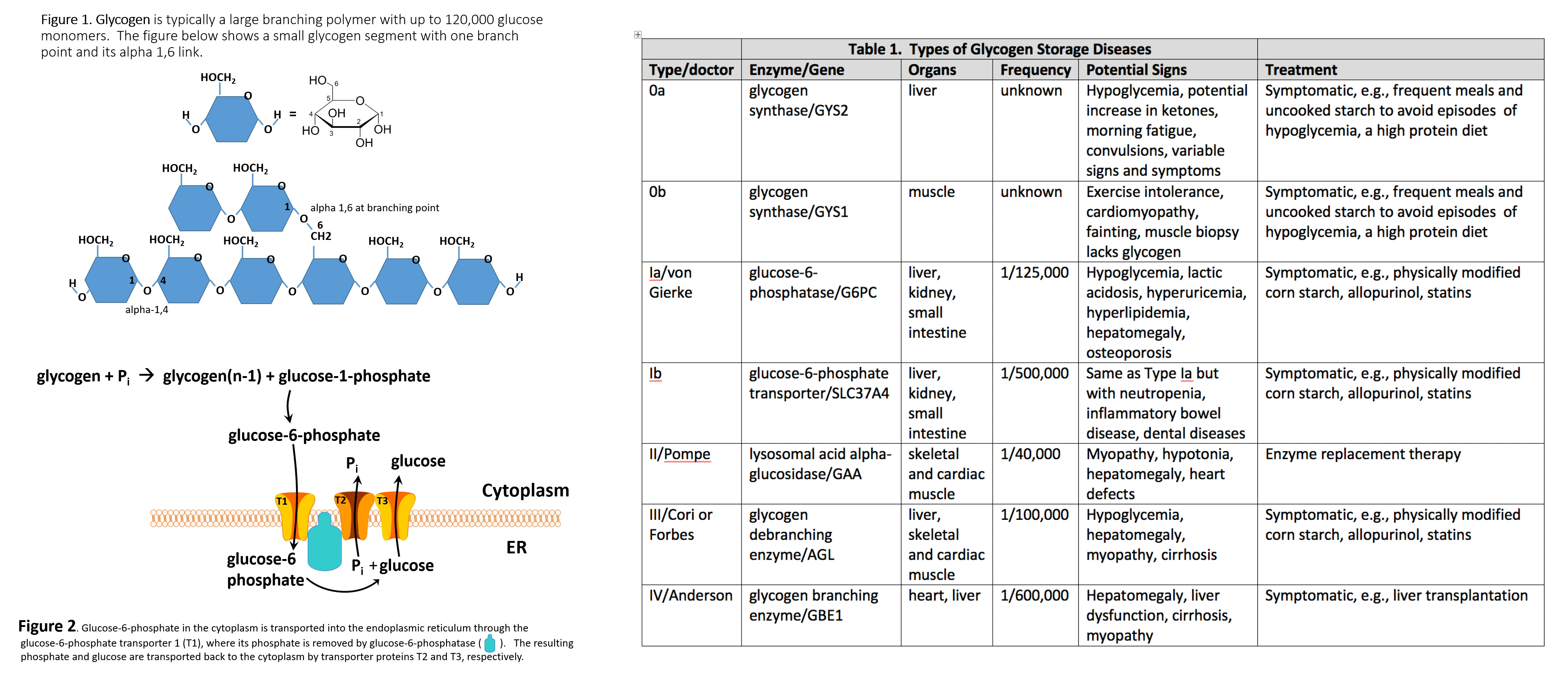
Glycogen Branching Polymer (left) Glycogen Storage Disease (right) Contributed by William Stone, MD
References
van den Hout HM, Hop W, van Diggelen OP, Smeitink JA, Smit GP, Poll-The BT, Bakker HD, Loonen MC, de Klerk JB, Reuser AJ, van der Ploeg AT. The natural course of infantile Pompe's disease: 20 original cases compared with 133 cases from the literature. Pediatrics. 2003 Aug:112(2):332-40 [PubMed PMID: 12897283]
Level 3 (low-level) evidenceKishnani PS, Corzo D, Leslie ND, Gruskin D, Van der Ploeg A, Clancy JP, Parini R, Morin G, Beck M, Bauer MS, Jokic M, Tsai CE, Tsai BW, Morgan C, O'Meara T, Richards S, Tsao EC, Mandel H. Early treatment with alglucosidase alpha prolongs long-term survival of infants with Pompe disease. Pediatric research. 2009 Sep:66(3):329-35. doi: 10.1203/PDR.0b013e3181b24e94. Epub [PubMed PMID: 19542901]
Level 1 (high-level) evidenceKishnani PS, Steiner RD, Bali D, Berger K, Byrne BJ, Case LE, Crowley JF, Downs S, Howell RR, Kravitz RM, Mackey J, Marsden D, Martins AM, Millington DS, Nicolino M, O'Grady G, Patterson MC, Rapoport DM, Slonim A, Spencer CT, Tifft CJ, Watson MS. Pompe disease diagnosis and management guideline. Genetics in medicine : official journal of the American College of Medical Genetics. 2006 May:8(5):267-88 [PubMed PMID: 16702877]
Level 3 (low-level) evidenceWinkel LP, Hagemans ML, van Doorn PA, Loonen MC, Hop WJ, Reuser AJ, van der Ploeg AT. The natural course of non-classic Pompe's disease; a review of 225 published cases. Journal of neurology. 2005 Aug:252(8):875-84 [PubMed PMID: 16133732]
Level 3 (low-level) evidenceKishnani PS, Hwu WL, Mandel H, Nicolino M, Yong F, Corzo D, Infantile-Onset Pompe Disease Natural History Study Group. A retrospective, multinational, multicenter study on the natural history of infantile-onset Pompe disease. The Journal of pediatrics. 2006 May:148(5):671-676 [PubMed PMID: 16737883]
Level 2 (mid-level) evidenceLaforêt P, Nicolino M, Eymard PB, Puech JP, Caillaud C, Poenaru L, Fardeau M. Juvenile and adult-onset acid maltase deficiency in France: genotype-phenotype correlation. Neurology. 2000 Oct 24:55(8):1122-8 [PubMed PMID: 11071489]
Pompe Disease Diagnostic Working Group, Winchester B, Bali D, Bodamer OA, Caillaud C, Christensen E, Cooper A, Cupler E, Deschauer M, Fumić K, Jackson M, Kishnani P, Lacerda L, Ledvinová J, Lugowska A, Lukacs Z, Maire I, Mandel H, Mengel E, Müller-Felber W, Piraud M, Reuser A, Rupar T, Sinigerska I, Szlago M, Verheijen F, van Diggelen OP, Wuyts B, Zakharova E, Keutzer J. Methods for a prompt and reliable laboratory diagnosis of Pompe disease: report from an international consensus meeting. Molecular genetics and metabolism. 2008 Mar:93(3):275-81 [PubMed PMID: 18078773]
Level 3 (low-level) evidenceSmith BK, Martin AD, Lawson LA, Vernot V, Marcus J, Islam S, Shafi N, Corti M, Collins SW, Byrne BJ. Inspiratory muscle conditioning exercise and diaphragm gene therapy in Pompe disease: Clinical evidence of respiratory plasticity. Experimental neurology. 2017 Jan:287(Pt 2):216-224. doi: 10.1016/j.expneurol.2016.07.013. Epub 2016 Jul 21 [PubMed PMID: 27453480]