Introduction
Holoprosencephaly (HPE) results from an incomplete midline cleavage of the forebrain (prosencephalon). It includes a wide spectrum of intracranial and craniofacial midline defects and a myriad of clinical manifestations, consisting of neurologic impairment and dysmorphism of the brain and face. It is the most common malformation of forebrain development.[1] Evidence suggests that HPE can be present either sporadically or can have a syndromic association. The defect associated with HPE occurs at approximately 3 to 4 weeks post-conception (between day 18 and day 28 of embryonic life) and is a disorder of gastrulation.[2]
The HPE phenotype continuum has been divided into 3 categories by DeMyer and Zeman and modified by DeMyer.[3][4] These categories include:
Alobar: This is the most severe and most common form (two-thirds of all HPE cases), characterized by a complete failure to partition the forebrain into the left and right hemispheres; this results in a single, centrally located ventricle.
Semilobar: Partial forebrain cleavage
Lobar: Almost complete forebrain cleavage
Middle interhemispheric variant (syntelencephaly): A rare variant with an abnormal midline union of the posterior frontal and parietal lobes.[5]
Septo-preoptic variant: This rarest variant is mild, and the midline fusion is restricted to the telencephalon's septal or preoptic region.[6]
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
Extensively researched cases of HPE primarily focus on individuals with normal karyotypes but with chromosomal anomalies, and these cases seldom coincide with malformation syndromes. In most live births affected by HPE, the condition is classified as nonsyndromic and nonchromosomal, indicating that it cannot be ascribed to syndromic or chromosomal origins. This category encompasses environmental causes, deletions, or gene mutations that play a role in HPE or unidentified factors.
The etiology of HPE can be divided into several categories, as follows:
Genetic Causes
Nonsyndromic monogenic causes: SHH anomalies are the most common defects recorded in HPE patients with associated midline defects. Other leading genetic causes include ZIC2, SIX3, and TGIF.[7]
The main functions of these genes include the following:
- Zinc finger protein (ZIC2) - Arrangement of the anterior notochord
- SIX homeobox 3 (SIX3 ) - Arrangement of the anterior-posterior polarity of brain vesicles
- Sonic hedgehog (SHH) - Telencephalic vesicle and optic cup patterning from the neural tube's ventral midline section
- Bone morphogenetic protein - Craniofacial patterning
- Fibroblast growth factor - Craniofacial patterning
- TG interacting factors (Tgifs) 1 and 2 - Regulation of SHH signaling [8]
Syndromic causes: Approximately 32% to 42% of the cases occur due to anomalies of chromosome number, most commonly trisomy 13 (Patau syndrome), followed by trisomy 18 and triploidy.[9] Patau syndrome is commonly caused by mosaicism, complete trisomy, or even by translocation of chromosome 13.[10] Almost 24% to 45% of these patients have associated HPE.[11]
Specific syndromes related to HPE include both autosomal dominant and autosomal recessive syndromes, including:
- Autosomal Dominant:
- CDON gene-related Steinfield syndrome
- FGFR1 gene-related Kallman syndrome and Hartsfield syndrome (holoprosencephaly, cleft lip and palate, and ectrodactyly)
- Agnathia-Otocephaly Complex (gene PRRX1)
- Culler-Jones syndrome
- CHARGE syndrome
- Autosomal Recessive:
- CENPF gene-related Stromme syndrome
- DHCR7 gene-related Smith-Lemli-Opitz Syndrome - intellectual disability, dysmorphisms of the face, congenital cardiac anomalies, and male external genitalia defects [12][13]
- HHAT (encoding Hedgehog acyltransferase)
- PLCH1 (encoding phospholipase C eta-1)
- STIL [encoding a factor required for maintenance of primary cilia) [3]
- Hydrolethalus syndrome (HYLS1 and KIF7 genes)
- Genoa syndrome
- Lambotte syndrome
- X-linked:
- X-linked holoprosencephaly-13 (HPE13) (STAG2) [14]
- Variants in genes:
- Driver genes: Essential to the phenotype of the patient carrying a variant-SHH, ZIC2, SIX3, FGF8, and FGFR1
- Silent modifier gene: BOC
- Predisposing genes: All other genes responsible
Nongenetic Causes
HPE exhibits associations with various maternal factors. Pregestational maternal diabetes is 1 such factor. Additionally, maintaining consistent maternal folic acid supplementation appears to offer protective benefits. Studies in animals have provided insights into potential teratogenic influences leading to HPE, including exposure to substances like ethanol, salicylate, retinoic acid, mycotoxins such as ochratoxins, Hedgehog signaling inhibitors like cyclopamine, and drugs that disrupt cholesterol biosynthesis.[15] Furthermore, environmental factors, including polycyclic aromatic hydrocarbons from sources like cigarette smoke and charred meats, Δ9-tetrahydrocannabinol (THC) found in cannabis, and piperonyl butoxide (PBO), a pesticide synergist frequently detected in indoor dust, also warrant consideration in understanding HPE's etiology.[3]
Epidemiology
HPE manifests relatively frequently, with an occurrence rate of approximately 1 in 250 conceptuses. However, the condition's birth prevalence, ranging from 1 in 8000 to 1 in 16,000 live births, remains consistent across diverse international populations. Within the United States, certain ethnic groups, including African-American, Hispanic, and Pakistani communities, appear to exhibit slightly elevated prevalence rates. This phenomenon is likely due to reduced rates of prenatal diagnosis and associated terminations within these particular groups.[2][16]
Pathophysiology
Understanding the relationship between the development of the face and the brain to comprehend the pathogenesis of HPE is essential. The successful development of the brain and face occurs due to the reciprocal signaling of several molecules between the surface ectoderm and neural crest cells that determine the migration of primitive embryonal cells, leading to the formation of highly developed structures. Incomplete diverticulation and cleavage of the embryonic forebrain is the reason for the intracranial pathology. Blocking this signaling alters the dorsoventral polarity and anterior-posterior axis and inhibits bilateral symmetry of the forebrain. SHH is 1 such signaling molecule. Inhibiting SHH expression in the basal telencephalon subsequently impedes the dorsoventral polarity and inhibits the induction of SHH expression in the frontonasal ectodermal zone (FEZ), leading to multiple midline facial defects. Various degrees of a block in the Shh signaling determine the severity of the disease manifestation.
Incomplete penetrance and variable expressivity make predicting disease manifestation in offspring difficult. Parents carrying defined mutations may not have either physical or neurological symptoms. Current studies suggest a multi-hit origin of HPE, explaining the lack of clinical manifestations in patients with the concerned mutations.[17][18][19][20] Basal forebrain patterning occurs during the third and fourth week of embryogenesis. This directs the cleavage of the prosencephalon during the fifth and sixth weeks of gestation.[1]
History and Physical
A standardized diagnostic protocol, including dysmorphology examination, complete family history and ascertainment of risk factors, and neuroimaging, covers these disorders' presenting features. The pathogenesis of HPE is related to abnormal genes responsible for the development of the head and the face. Hence, clinically, the spectrum of the presentation can range from mild craniofacial defects, like a single maxillary central incisor with normal neurological function, to severe cranio-facial-neurological defects, like cyclopia or proboscis with an alobar variant. In moderate to severe cases, craniofacial symptoms have proven to indicate the severity of the intracranial lesion.
Severe facial phenotypes are associated with the alobar variant of HPE and include the following:
- Pronounced microcephaly
- Cyclopia: Single, centrally placed eye, synophthalmia, or anophthalmia. Proboscis (a tube-like nasal appendage with a single nostril above the ocular region) may be single or absent and may or may not be accompanied by hypognathism
- Cebocephaly: A combination of ocular hypotelorism and a single-nostril nose
- Ethmocephaly: Proboscis with ocular hypotelorism [21]
Less severe facial phenotypes are associated with intracranial manifestations as follows:
- Hypotelorism is associated with the lobar variant.
- The lobar variant is associated with midface hypoplasia, a flat nasal bridge, cleft lip and/or palate, and agenesis of the columella.
- Ocular colobomas and other severe features are related to the alobar variant (while when present alone, they are a sign of the lobar/middle interhemispheric variant).
- The single maxillary central incisor-microform variant is also a less severe phenotype.
Individuals with a mild variant of HPE usually have a relative with frank HPE. These patients typically present with only craniofacial anomalies without the accompanying neurological defect.[2]
Evaluation
In a pediatric patient fulfilling the clinical triad of atypical morphology, family history, and environmental factors, the next step is to confirm the type of intracranial anomaly by neuroimaging. Evaluation of HPE can take place as early as the prenatal period. Noninvasive prenatal testing (NIPT) is a useful method for diagnosing fetal trisomy 13 in the first trimester.[22] The evaluation includes ultrasonography (USG) and magnetic resonance imaging (MRI). In severe forms of HPE, a USG can detect the anomaly as early as the first trimester. An MRI is used for diagnosis in the third trimester. Postnatal neuroimaging includes USG, the preferred imaging modality in infants with open anterior fontanelles. A computed tomography (CT) or MRI may be considered in older children. However, testing comes with risks. While the CT can cause excess radiation exposure, the risk of excess sedation exists with MRI.
Patterns observed on imaging include the following:
- Alobar HPE: This occurs due to diffuse cortical non-separation. The corpus callosum and olfactory bulbs are absent entirely. The deep gray nuclei are fused, and a single midline ventricle is visualized in these patients (see Image. Alobar Holoprosencephaly).
- Semilobar form: The frontal lobes are non-separated, and the anterior corpus callosum is absent. Fused deep gray nuclei and absent anterior horns of lateral ventricles are characteristic. The posterior horns of the lateral ventricles are well-developed.
- Lobar form: This defect is associated with the non-separation of basal frontal brains, the absence of the corpus callosum adjacent to affected areas with hypoplastic olfactory bulbs, and the azygous anterior cerebral artery (anteriorly displaced).
- Middle interhemispheric variant (syntelencephaly): In these patients, nondisjunction between bilateral frontal and parietal lobes, an absent body of the corpus callosum, and an azygous anterior cerebral artery are evident.[2] The frontal horns are better developed than in alobar HPE, but the occipital horns are poorly developed and are seen as a single ventricular cavity.
- Microform HPE: There are no brain malformations in this variant. However, facial anomalies can be seen that include a single maxillary incisor, clefting, hypotelorism, and retinal coloboma.[23]
Signs on imaging include the following:
- A horse-shoe-shaped mono ventricle can be seen on imaging.
- Butterfly sign: This is the visualization of the normal appearance of both choroid plexes in the axial plane on an antenatal ultrasound scan. Its absence suggests HPE.
- Absence of cavum septi pellucidi: Individuals with all types of HPE lack a normal cavum septi pellucidi.
- “Snake under the skull” sign: The defective cortical tissue bridge between the 2 frontal gyri in lobar HPE pushes the anterior cerebral artery outside alongside the frontal bone.
Electroencephalography findings are as follows:
- Multifocal spikes that often evolve into hypoarrhythmia are found in EEG.
- The waking EEG in the neonatal period classically shows a continuous high-voltage alpha-theta monorhythmic activity that becomes discontinuous while the child sleeps.
Once the diagnosis of HPE is confirmed, the patient is evaluated for a syndromic association. If a syndromic association is discovered, the management course changes to include evaluating any coexisting features that might require further medical or surgical attention.
Genetic testing is carried out to determine the existence of any chromosomal anomaly by conducting cytogenetic and molecular testing. If this reveals any chromosomal or genetic anomaly associated with HPE, it further mandates genetic counseling for the parents should they decide to conceive another child.[24]
Treatment / Management
Managing HPE is symptomatic and requires a multidisciplinary approach. This condition targets each organ system and the related complications as follows:
- Epilepsy is a common complication in patients with HPE. A single seizure occurs in approximately 50% of all children with HPE, some of whom may require multidrug antiepileptic therapy.[13]
- When HPE is associated with macrocephaly, coexisting hydrocephalus is typically present; this should be immediately treated with shunt surgery while preventing over-drainage of cerebrospinal fluid (CSF).[25]
- Motor anomalies, when present, warrant treatment. Spasticity and dystonia require pharmacological interventions, including intrathecal baclofen pumps, oral trihexyphenidyl, and physical and occupational therapy.
- Orofacial motor dysfunction is compounded by structural anomalies like cleft palate/lip causing aspiration, further progressing to pneumonia, posing a serious feeding challenge; this can require structural surgical correction.
- The hormone dysregulation caused by the absence of a well-formed hypothalamus can manifest as pituitary hormone deficiencies, most commonly diabetes insipidus. The manifestation of other hormonal deficits is rare. However, prednisone, thyroxine, and growth hormone supplements may be required in patients with overt deficiency symptoms.
- Poor nerve migration to the gastrointestinal tract causes poor gastric and colonic motility and gastroesophageal reflux; this can be managed by placing a gastrostomy tube in severe facial defects. Medications and antireflux procedures in patients with isolated severe gastroesophageal reflux disease are indicated in other cases.[2] (B3)
Differential Diagnosis
When considering a diagnosis of HPE, it is crucial to explore a range of potential differential diagnoses, including the following:
Septo-Optic dysplasia: In addition to enlarged ventricles and hypothalamic-pituitary axis failure, this diagnosis also consists of optic nerve degeneration and absent septum pellucidum.[26]
DiGeorge syndrome: Abnormalities such as the cleft palate and hypertelorism are associated with other anomalies, including cardiopulmonary anomalies like aortic arch and conotruncal defects. Other anomalies like T-cell deficiency and hypocalcemia resulting from parathyroid hypoplasia are also seen.[27]
Hydranencephaly: A CT of the head shows absent bilateral cerebral hemispheres with no cortical mantle and a fluid-filled cavity. However, since the entire falx is preserved, HPE is excluded.[28]
Porencephalic cyst: On MRI, this finding appears as an abnormal accumulation of CSF within the brain parenchyma rather than an absence/midline fusion of cerebral lobes, as seen in HPE.[29]
Arachnoid cyst: This diagnosis occurs in conjunction with HPE or could be the only finding. Arachnoid cysts occur secondary to trauma or could be inherited in an autosomal manner. This condition does not present with facial defects unless coexisting HPE exists.[29][30]
Prognosis
Moderate to severe cases of HPE generally have a poor prognosis. The mortality rate increases with 33% of neonates dying in the first 24 hours after birth, 58% in the first month, 50% between the fourth and fifth months, and 70% to 80% in the first year of life. A very small number of these neonates survive until adulthood. However, a recent study from a huge population registry showed that the 1-year survival rate was 58.1% and the 10-year survival was 36.9% for HPE.[31] Mild to moderate cases typically survive to adulthood but live with complications.[13] About 50% of lobar HPE patients can ambulate and verbally communicate.[1]
Complications
Neurological complications associated with HPE include epilepsy with or without clinically evident seizure activity and motor impairment such as hypotonia and dystonia with or without spasticity.[18] Hydrocephalus is a common complication with the alobar variant of HPE, thus presenting with a macrocephaly rather than microcephaly, sometimes clouding the initial clinical suspicion.[25] Gastrointestinal tract anomalies like poor gastric and colonic motility and gastroesophageal reflux result from poor neuronal migration during development.[32]
The hypothalamus, a midline structure, is unformed in these patients. Non-formation of the hypothalamus is associated with various endocrinal symptoms due to pituitary hormone deficiency and impaired homeostatic functions like thirst, hunger, and temperature. Hormone irregularities of the posterior pituitary are far more common than those of the anterior pituitary. Patients must be screened repeatedly for electrolyte levels to diagnose diabetes insipidus. Regarding anterior pituitary insufficiency, both hypothyroidism and hypocortisolism can be lethal, while growth hormone and gonadotrophic hormone deficiencies can lead to stunting and sexual immaturity, respectively.[2]
Deterrence and Patient Education
Genetic testing is recommended for parents with children showing apparent symptoms or for those showing mild variants.[12] However, the indication for testing depends on the disease's clinical suspicion, which is very difficult given the numerous phenotypic variations. The number of genes involved and a multi-hit process compound the challenging diagnosis further. The de-novo occurrence of some mutations that show a specific defect in the parents has also been observed. In these cases, the chances of HPE in a subsequent pregnancy are significantly reduced.[24]
Pearls and Other Issues
Pertinent clinical points relevant to HPE are as follows:
- HPE occurs due to incomplete midline cleavage of the forebrain (prosencephalon) and includes a wide spectrum of intracranial and craniofacial midline defects.
- The etiology of HPE is genetic or sporadic. Genetic causes can be further classified into syndromic and nonsyndromic causes.
- The severity of craniofacial defects often correlates with the degree of the intracranial anomaly.
- A diagnosis can also be established prenatally using a USG in the first trimester and an MRI in the third trimester.
- An MRI is used to diagnose HPE definitively. Depending on the clinical and MRI findings, HPE can be classified into alobar, semilobar, lobar, and middle intrahemispheric variants.
- Most patients with HPE have associated seizures, hydrocephalus, motor dysfunction, endocrine dysfunction, or gastric dysmotility.
- The prognosis depends on the degree of an anomaly in the patient. While the alobar, lobar, and semilobar variants have a poorer prognosis, the middle intrahemispheric variant has a somewhat better prognosis.
- Genetic testing is recommended for parents with a child showing apparent symptoms or for those showing mild variants.
Enhancing Healthcare Team Outcomes
The complex nature of HPE and its associated complications require an interdisciplinary approach. This approach involves various healthcare professionals. Endocrine dysfunction that commonly accompanies some variants of HPE should be treated lifelong by a pediatric endocrinologist. Pediatric gastroenterologists attend to associated gastrointestinal anomalies. Neurological anomalies like seizures, movement disorders, facial anomalies, and hydrocephalus are best managed by a neurologist and a neurosurgeon. Dysphagia or reflux related to the cleft palate is treated by a plastic surgeon and a pediatric surgeon. The importance of psychological aid and genetic counseling must not be undermined and should be offered to all parents. Child psychiatrists and psychologists should be consulted for the emotional and behavioral consequences of HPE. Nurses, physical and occupational therapists, speech pathologists, and social workers assist in addressing the patient's physical, developmental, and emotional needs.
HPE is a complex condition with evolving research and treatment strategies. The earlier the signs and symptoms of an HPE complication are identified, the better the prognosis and outcome.[33] Collaboration, shared decision-making, and good communication among the interprofessional team are critical for optimal outcomes. The interprofessional care provided to the patient demands an integrated, comprehensive care pathway combined with an evidence-based approach. Healthcare professionals should stay updated through ongoing education and collaboration with academic institutions.
The clinical skills to treat HPE and a deep commitment to ethical decision-making, family support, and interprofessional collaboration enhance patient-centered care, patient safety, and team performance. This comprehensive strategy can ultimately lead to improved patient outcomes and a higher quality of life for individuals and families affected by HPE.
Media
(Click Image to Enlarge)
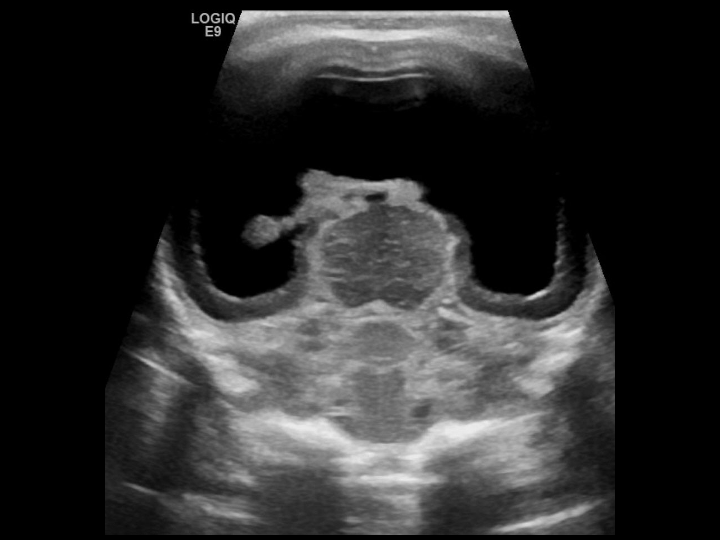
Alobar Holopresencephaly. Coronal gray-scale US image shows a midline monoventricle, fused thalami, thinned cerebral mantle and absent inter hemispheric fissure.
Contributed by PK Sandhu, MD
References
Riddle A, Nagaraj U, Hopkin RJ, Kline-Fath B, Venkatesan C. Fetal Magnetic Resonance Imaging (MRI) in Holoprosencephaly and Associations With Clinical Outcome: Implications for Fetal Counseling. Journal of child neurology. 2021 Apr:36(5):357-364. doi: 10.1177/0883073820972290. Epub 2020 Nov 23 [PubMed PMID: 33226281]
Level 2 (mid-level) evidenceRaam MS, Solomon BD, Muenke M. Holoprosencephaly: a guide to diagnosis and clinical management. Indian pediatrics. 2011 Jun:48(6):457-66 [PubMed PMID: 21743112]
Lo HF, Hong M, Krauss RS. Concepts in Multifactorial Etiology of Developmental Disorders: Gene-Gene and Gene-Environment Interactions in Holoprosencephaly. Frontiers in cell and developmental biology. 2021:9():795194. doi: 10.3389/fcell.2021.795194. Epub 2021 Dec 22 [PubMed PMID: 35004690]
DEMYER W, ZEMAN W, PALMER CG. THE FACE PREDICTS THE BRAIN: DIAGNOSTIC SIGNIFICANCE OF MEDIAN FACIAL ANOMALIES FOR HOLOPROSENCEPHALY (ARHINENCEPHALY). Pediatrics. 1964 Aug:34():256-63 [PubMed PMID: 14211086]
Rajalakshmi PP, Gadodia A, Priyatharshini P. Middle interhemispheric variant of holoprosencephaly: A rare midline malformation. Journal of pediatric neurosciences. 2015 Jul-Sep:10(3):244-6. doi: 10.4103/1817-1745.165678. Epub [PubMed PMID: 26557166]
Hahn JS, Barnes PD, Clegg NJ, Stashinko EE. Septopreoptic holoprosencephaly: a mild subtype associated with midline craniofacial anomalies. AJNR. American journal of neuroradiology. 2010 Oct:31(9):1596-601. doi: 10.3174/ajnr.A2123. Epub 2010 May 20 [PubMed PMID: 20488907]
Srivastava K, Hu P, Solomon BD, Ming JE, Roessler E, Muenke M. Molecular analysis of the Noggin (NOG) gene in holoprosencephaly patients. Molecular genetics and metabolism. 2012 Jun:106(2):241-3. doi: 10.1016/j.ymgme.2012.03.008. Epub 2012 Mar 21 [PubMed PMID: 22503063]
Taruscio D,Mantovani A, The rare malformation holoprosencephaly: pathogenesis, association with pregestational diabetes and the possible link with food pollutants. Annali dell [PubMed PMID: 38088397]
Kruszka P, Muenke M. Syndromes associated with holoprosencephaly. American journal of medical genetics. Part C, Seminars in medical genetics. 2018 Jun:178(2):229-237. doi: 10.1002/ajmg.c.31620. Epub 2018 May 17 [PubMed PMID: 29770994]
Williams GM, Brady R. Patau Syndrome. StatPearls. 2024 Jan:(): [PubMed PMID: 30855930]
Costa AD, Schultz R, Rosemberg S. Alobar holoprosencephaly and Trisomy 13 (Patau syndrome). Autopsy & case reports. 2013 Apr-Jun:3(2):5-10. doi: 10.4322/acr.2013.012. Epub 2013 Jun 30 [PubMed PMID: 31528602]
Level 3 (low-level) evidenceAdam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, Tekendo-Ngongang C, Muenke M, Kruszka P. Holoprosencephaly Overview. GeneReviews(®). 1993:(): [PubMed PMID: 20301702]
Level 3 (low-level) evidenceHoney EM, Bütow KW, Zwahlen RA. Holoprosencephaly with Clefts: Data of 85 Patients, Treatment and Outcome: Part 1: History, Subdivisions, and Data on 85 Holoprosencephalic Cleft Patients. Annals of maxillofacial surgery. 2019 Jan-Jun:9(1):140-145. doi: 10.4103/ams.ams_50_19. Epub [PubMed PMID: 31293943]
Kruszka P, Berger SI, Casa V, Dekker MR, Gaesser J, Weiss K, Martinez AF, Murdock DR, Louie RJ, Prijoles EJ, Lichty AW, Brouwer OF, Zonneveld-Huijssoon E, Stephan MJ, Hogue J, Hu P, Tanima-Nagai M, Everson JL, Prasad C, Cereda A, Iascone M, Schreiber A, Zurcher V, Corsten-Janssen N, Escobar L, Clegg NJ, Delgado MR, Hajirnis O, Balasubramanian M, Kayserili H, Deardorff M, Poot RA, Wendt KS, Lipinski RJ, Muenke M. Cohesin complex-associated holoprosencephaly. Brain : a journal of neurology. 2019 Sep 1:142(9):2631-2643. doi: 10.1093/brain/awz210. Epub [PubMed PMID: 31334757]
Kauvar EF, Muenke M. Holoprosencephaly: recommendations for diagnosis and management. Current opinion in pediatrics. 2010 Dec:22(6):687-95. doi: 10.1097/MOP.0b013e32833f56d5. Epub [PubMed PMID: 20859208]
Level 3 (low-level) evidenceGeng X, Oliver G. Pathogenesis of holoprosencephaly. The Journal of clinical investigation. 2009 Jun:119(6):1403-13. doi: 10.1172/JCI38937. Epub 2009 Jun 1 [PubMed PMID: 19487816]
Level 3 (low-level) evidencePetryk A, Graf D, Marcucio R. Holoprosencephaly: signaling interactions between the brain and the face, the environment and the genes, and the phenotypic variability in animal models and humans. Wiley interdisciplinary reviews. Developmental biology. 2015 Jan-Feb:4(1):17-32. doi: 10.1002/wdev.161. Epub 2014 Oct 22 [PubMed PMID: 25339593]
Level 3 (low-level) evidenceMing JE, Muenke M. Multiple hits during early embryonic development: digenic diseases and holoprosencephaly. American journal of human genetics. 2002 Nov:71(5):1017-32 [PubMed PMID: 12395298]
Level 3 (low-level) evidenceKruszka P, Martinez AF, Muenke M. Molecular testing in holoprosencephaly. American journal of medical genetics. Part C, Seminars in medical genetics. 2018 Jun:178(2):187-193. doi: 10.1002/ajmg.c.31617. Epub 2018 May 17 [PubMed PMID: 29771000]
Golden JA. Towards a greater understanding of the pathogenesis of holoprosencephaly. Brain & development. 1999 Dec:21(8):513-21 [PubMed PMID: 10598051]
Level 3 (low-level) evidenceAruna E, Chakravarthy VK, Rao DN, Rao DR. Holoprosencephaly with multiple anomalies of the craniofacial bones-an autopsy report. Journal of clinical and diagnostic research : JCDR. 2013 Aug:7(8):1722-4. doi: 10.7860/JCDR/2013/5734.3268. Epub 2013 Aug 1 [PubMed PMID: 24086891]
Chen CP. Positive non-invasive prenatal testing for trisomy 13 in the first trimester in a pregnancy with fetal holoprosencephaly, cebocephaly and postaxial polydactyly. Taiwanese journal of obstetrics & gynecology. 2024 Jan:63(1):105-107. doi: 10.1016/j.tjog.2023.10.007. Epub [PubMed PMID: 38216244]
Addissie YA, Troia A, Wong ZC, Everson JL, Kozel BA, Muenke M, Lipinski RJ, Malecki KMC, Kruszka P. Identifying environmental risk factors and gene-environment interactions in holoprosencephaly. Birth defects research. 2021 Jan 1:113(1):63-76. doi: 10.1002/bdr2.1834. Epub 2020 Oct 28 [PubMed PMID: 33111505]
Dubourg C, Bendavid C, Pasquier L, Henry C, Odent S, David V. Holoprosencephaly. Orphanet journal of rare diseases. 2007 Feb 2:2():8 [PubMed PMID: 17274816]
Level 3 (low-level) evidenceSarica C, Yucetas C, Ozen A, Ucler N, Konca C, Akar S. Management Strategies for Hydrocephalus in Alobar Holoprosencephaly: A Case Report and Discussion. Pediatric neurosurgery. 2018:53(5):337-341. doi: 10.1159/000489856. Epub 2018 Jun 14 [PubMed PMID: 29902800]
Level 3 (low-level) evidenceMaurya VK, Ravikumar R, Bhatia M, Rai R. Septo-optic dysplasia: Magnetic Resonance Imaging findings. Medical journal, Armed Forces India. 2015 Jul:71(3):287-9. doi: 10.1016/j.mjafi.2015.04.013. Epub 2015 Jun 29 [PubMed PMID: 26286097]
Lackey AE, Muzio MR. DiGeorge Syndrome. StatPearls. 2024 Jan:(): [PubMed PMID: 31747205]
Khalid M, Khalid S, Zaheer S, Redhu N, Ekramullah. Hydranencephaly: a rare cause of an enlarging head size in an infant. North American journal of medical sciences. 2012 Oct:4(10):520-2. doi: 10.4103/1947-2714.102015. Epub [PubMed PMID: 23112982]
El Hasbani G, Balaghi A, Assaker R, Rojas A, Troya M, Kofahi A, Assaker JP, Diab C, Al Husayni H. Intraparenchymal hemorrhage and cerebral venous thrombosis in an adult with congenital porencephalic cyst presenting for generalized tonic-clonic seizures. Radiology case reports. 2020 Jan:15(1):95-99. doi: 10.1016/j.radcr.2019.10.028. Epub 2019 Nov 12 [PubMed PMID: 31762865]
Level 3 (low-level) evidenceXiong J, Xiang B, Chen X, Cai T. Case report: a novel mutation in ZIC2 in an infant with microcephaly, holoprosencephaly, and arachnoid cyst. Medicine. 2019 Mar:98(10):e14780. doi: 10.1097/MD.0000000000014780. Epub [PubMed PMID: 30855487]
Level 3 (low-level) evidenceBenjamin RH, Nguyen JM, Canfield MA, Shumate CJ, Agopian AJ. Survival of neonates, infants, and children with birth defects: a population-based study in Texas, 1999-2018. Lancet regional health. Americas. 2023 Nov:27():100617. doi: 10.1016/j.lana.2023.100617. Epub 2023 Oct 18 [PubMed PMID: 37868647]
Levey EB, Stashinko E, Clegg NJ, Delgado MR. Management of children with holoprosencephaly. American journal of medical genetics. Part C, Seminars in medical genetics. 2010 Feb 15:154C(1):183-90. doi: 10.1002/ajmg.c.30254. Epub [PubMed PMID: 20104615]
Buljac-Samardzic M, Doekhie KD, van Wijngaarden JDH. Interventions to improve team effectiveness within health care: a systematic review of the past decade. Human resources for health. 2020 Jan 8:18(1):2. doi: 10.1186/s12960-019-0411-3. Epub 2020 Jan 8 [PubMed PMID: 31915007]
Level 1 (high-level) evidence