Introduction
Kawasaki disease (KD), also known by the name mucocutaneous lymph node syndrome, is an acute, self-limited medium vessel vasculitis that has a predilection for the coronary arteries.[1] It is the leading cause of acquired heart disease in developed nations and is slowly bypassing rheumatic heart disease in developing countries.[2][3]
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
The etiology of Kawasaki disease (KD) is still not known and may be related to a wind-borne or water-borne pathogen. However, in recent years there have been studies that have shown certain genetic markers (such as HLA-B51 and HLA-Bw22j2 serotypes, chemokine receptor gene-cluster CCR2-CCR5 haplotypes and FCGR3A polymorphism of the IgG receptor IIIa) show a predisposition to the disease. In fact, siblings are 10-20 times more likely to develop the disease when compared to the general population.[1][3][4] There is no evidence to suggest that the disease passes from person to person. Multiple organisms of bacterial and viral origin have are under suspicion as a cause, but to date, no single agent has been identified as the leading cause.[1] However, there has been evidence showing over 40% of children diagnosed with KD have tested positive for viral respiratory pathogens.[5]
Epidemiology
This disease is most common in children younger than five years of age, but it can present at any age, even in adults.[4] There is a slight male predominance (male to female = 1.5 to 1).[2][6][4] Boys are also more likely to suffer from complications and death.[3] It is rare to see Kawasaki disease in children less than 4 months, possibly pointing to protection from maternal antibodies.[1] It is most commonly seen in children of Asian descent, particularly Japanese, and is least common in Caucasian children.[1] There also appears to be a higher prevalence of the disease in the winter and spring months.[1][3][4] Incidence varies from 10 to 20 in 100,000 children aged < 5 years in the U.S. and Canada to 50 to 250 in 100,000 in Japan, Taiwan or Korea.
Pathophysiology
An agent, most likely infectious, enters via the respiratory tract and starts a response that activates lymphocytes, cytokines, and proteinases, specifically tumor necrosis factor alpha (TNF-a), Interleukin 1, 4 and 6 and matrix metalloproteinases (MMP3 and MMP9)[7]. Oligoclonal IgA plasma cells are believed to play a crucial role in the cascade leading to coronary arteritis and are also prominent in the respiratory tract which is why the thinking is that a respiratory infection triggers the disease process.[1] This activation causes myocarditis and arteritis and, from fibrinoid necrosis of the internal elastic lamina, the development of a weak spot in the vessel wall that predisposes it to an aneurysmal formation from neutrophil invasion.[1] Atypical activation of monocytes and macrophages is considered the cause of these vascular lesions, which is unique to Kawasaki disease and is suggestive that an innate immune response triggers this process.[6][7][8] Over weeks and months, wall thickening of the coronary aneurysms can lead to stenosis and thrombus formation which can result in myocardial infarction (MI), rupture, ischemia-related dysrhythmias or death. The greatest risk of these cardiac complications is during the period of thrombocytosis. Small coronary aneurysms may resolve in 60% of cases in the later convalescent phase when inflammatory markers return to normal.
Histopathology
A biopsy is rarely performed and is not necessary to make a diagnosis. Findings early in the disease show destruction of the media of vessels by neutrophils.[8] Coronary arteritis characteristically presents with granulomatous inflammation that develops 6-8 days into the illness and is present across all layers of the vessel.[4][9] As the disease progresses, the infiltration gets replaced by lymphocytes, monocytes, and fibroblasts which lead to arterial remodeling.[8] Both light and electron microscopy show cytoplasmic inclusion bodies containing RNA in 85% of acute and late-stage fatalities.[10]
History and Physical
These children will present with multiple days of fever and generalized malaise. On physical exam, one will often find one or more of the diagnostic criteria listed below. It is important to get a full history because meeting any of the diagnostic criteria at any point during the illness count toward the diagnosis of Kawasaki disease, even if not present at the time of evaluation. There are three principal stages of the disease process: acute, subacute, and convalescent.[11][12] The acute phase consists of an abrupt onset of high fever that lasts 1 to 2 weeks but can last up to 3 to 4 weeks if left untreated. During the acute phase rash, conjunctivitis and myocarditis occur. The subacute phase begins when the fever subsides and lasts into weeks 4 to 6 of the disease course. The patient will have desquamation of the hands, thrombocytosis, and the development of coronary artery aneurysms. The convalescent phase is when the clinical signs of the illness cease, which is typically within 3 months from the initial onset of the disease. Cardiac abnormalities can still be apparent in this stage; however, new aneurysms after 8 weeks of illness are unusual.[13]
A variety of less common manifestations include abdominal pain, vomiting or diarrhea in 20%; hepatitis; parotitis; intussusception; joint pain in between 15-50%, especially the larger weight-bearing joints [1]; headache, irritability, seizure, aseptic meningitis; rhinorrhea and cough in 20-30%; aortic aneurysms; valvular insufficiency; epididymitis, orchitis or urethritis. Purulent conjunctivitis or exudative pharyngitis findings would each suggest a diagnosis other than KD.
In 2014, the American Heart Association (AHA) published the criteria needed to establish a diagnosis.[1] However, it is important to note that children who fall short of the full criteria but have cardiac abnormalities on echocardiogram meet the diagnosis of KD.[1] The patient must have fevers for five or more days, with at least four of the following criteria (either all at once or over a series of days):
- Bilateral painless bulbar conjunctival injection without exudate
- Erythematous mouth and pharynx, strawberry tongue or red, cracked lips
- Polymorphous exanthem (morbilliform, maculopapular, or scarlatiniform)
- Swelling of hands and feet with erythema of the palms and soles
- Cervical lymphadenopathy (over 1.5 cm in diameter)
Patients will typically have a patchy generalized macular erythematous rash over the trunk and extremities that appears within 5 days of the fever, and mocks a viral exanthem or drug eruption, but will lack pruritus.[1][3][14] Patients will also typically have peeling of the skin in the periungual area that starts about 2 to 3 weeks after onset of fever, which is characteristic of KD.[1] Nail changes, typically transverse depressions (Beau's lines), are seen in over 75% of patients with the disease that appear about 5 to 8 days after the onset of the fever and can last 2 to 4 weeks.[1][3][6]
Evaluation
The diagnosis of Kawasaki disease is clinical. You can use the mnemonic "Warm CREAM" or "FEBRILE" to help remember the criteria (see Table). The fever is the most consistent presentation, is minimally responsive to antipyretics, and typically remains higher than 38.5 degrees Celsius.[1][6] A conjunctival injection can also present with photophobia and can correlate with uveitis in about 65% of patients.[6] All of these symptoms tend to present sequentially, which indicates the diagnosis of KD over other disease processes on the differential.[2] The diagnosis of complete KD should be made in the acute phase of the process.[3]
Table: Warm CREAM and FEBRILE Mnemonics for KD Criteria
Warm = Fever for > 5 days
- C = Conjunctivitis without exudate
- R = Rash
- E = Edema or erythema of hands or feet, followed by desquamation and nail changes
- A = Adenopathy, often unilateral, cervical node > 1.5 cm
- M = Mucosal erythema, fissures or crusting of lips or strawberry tongue
- F = Fever for > 5 days
- E = Enanthem of mucosal membranes
- B = Bulbar conjunctivitis
- R = Rash, erythematous, polymorphous
- I = Internal organ involvement: coronary, abdominal, pneumonitis, hepatitis, orchitis
- E = Extremity changes, initial edema, and erythema, desquamation, nail changes
Incomplete Kawasaki disease should be a consideration in children with fevers of five or more days and two or three of the major criteria. It is more common in younger infants and older children.[1][2][4] Therefore, if an infant younger than 6 months has a prolonged fever of over 7 days, it is important to get an echocardiogram to rule out KD.[2][4] The AHA has created an algorithm for these patients which involves lab studies and an echocardiogram to make the diagnosis.[3] If a C-reactive protein (CRP) is < 30 mg/l and erythrocyte sedimentation rate (ESR) < 40 mm/hr, then follow the patient daily; if fever resolves and peeling of the skin occurs, get an echocardiogram and treat if aneurysms detected. If the CRP > 30 mg/l and/or ESR > 40 mm/hr, then get an echocardiogram. To meet incomplete KD criteria, initiate treatment, and obtain an echocardiogram, more than three of the following must be present[15]:
- Hemoglobin low for age
- White blood count (WBC) > 15,000/mm
- Platelet count > 450,000/mm
- Serum albumin < 3.0 g/dl
- Elevated alanine aminotransferase
- Urine WBC > 10/high-power field
There are no other required laboratory or diagnostic imaging studies necessary after a clinical diagnosis of KD, except an echocardiogram. However, some studies may be useful in the case of incomplete disease in an attempt to help narrow the diagnosis when not meeting all of the clinical criteria. Suggested studies include complete blood count (CBC), metabolic panel, ESR, CRP, and urinalysis. Mild-moderate normocytic anemia is characteristic of the acute disease phase, while thrombocytosis is present in the subacute phase.[6] Thrombocytopenia may be present less commonly in the acute phase, but predicts a greater risk for coronary artery aneurysm (CAA), and may be associated with immune thrombocytopenic purpura (ITP), disseminated intravascular coagulation (DIC), or destruction by immunoglobulin. Hypoalbuminemia may be present and correlates with a more severe and prolonged disease course.[8] Hyperbilirubinemia and elevated liver enzymes can be caused by hepatic congestion, which in turn can lead to obstructive jaundice as well as gallbladder hydrops.[1][6][16][17] Acute phase reactants are almost always elevated. Pro-B-type natriuretic peptide (Pro-BNP) levels appear to correlate with a greater risk of coronary artery aneurysm formation and intravenous immunoglobulin (IVIG) resistance in patients younger than 3 months old.[3] Urinalysis will show sterile pyuria, which will be absent if the sample was from from bladder aspiration.[1][6] If a lumbar puncture is performed to help rule out other differentials, pleocytosis is a common finding in KD and can be misleading to a diagnosis or viral meningitis [1]. Since concomitant respiratory viral infections are common and may play a role in pathogenesis in KD, do not let a positive respiratory viral polymerase chain reaction (PCR) finding mislead away from the diagnosis of KD.
If obtained, blood cultures, stool cultures, antinuclear antibody (ANA), rheumatoid factor (RF) and anti-streptolysin O (ASO) titers would be negative in KD cases.
An echocardiogram should be performed during the acute phase of the disease to rule out coronary artery aneurysm (CAA). The proximal left anterior descending and right coronary arteries are most commonly affected.[18] Aneurysms can be fusiform, saccular, ectatic, or segmented. Aneurysms more likely to regress are fusiform, small and distal. It is also important to evaluate other parts of the heart to look for signs of aortic root dilation, decreased contractility, valvular dysfunction, and effusion. However, ultrasound is subject to operator ability and has difficulty assessing the mid and distal portion of the vessels. Because of this, the use of CTA of the coronary arteries is becoming more prominent as it can identify and quantify lesions with precision, especially those in the mid and distal segments of the vessels.[19] However, one must weigh the risks versus the benefits of radiation exposure in the pediatric population. Also, CT is limited in evaluating the lumen of vessels that have calcification present.[4] Some investigators are now using electron beam CT to assess for coronary artery calcifications as a marker for future coronary events.[20] Magnetic resonance angiography (MRA) is also an option if radiation exposure is a concern, and it is more sensitive for small lesions and intimal hyperplasia.[4] Electrocardiography (ECG) can show a prolonged PR interval, deep Q waves, low voltage, ST-T changes, and arrhythmias which are suggestive of myocardial damage and repolarization abnormalities.[4]
Cardiac stress testing for reversible ischemia has been recommended as Level A for children with KD and known CAAs.[1] Coronary angiography offers more detailed views of the coronary arteries when compared to echocardiogram, but is only of benefit in patients who have more complex coronary artery lesions to assess for stenosis.[1][4] It is recommended to delay angiography 6-12 months after the initial presentation.[1]
Treatment / Management
Treatment aims with Kawasaki disease point to minimize the risk of coronary artery aneurysm (CAA) formation, which peaks two to four weeks after illness onset, by decreasing the inflammation of the coronary arteries.[4] Supportive care is also essential. Patients should receive high dose IVIG at 2 g/kg over 10-12 hours as well as high dose aspirin (ASA) (80 mg/kg/day to 100 mg/kg/day divided every six hours) until the patient has been afebrile for over 48 hours.[1] At that time ASA should be continued, but the dosing can be decreased to 3 mg/kg-5 mg/kg and maintained until there is no longer any evidence of cardiac changes about 6 to 8 weeks after illness onset.[1] ASA is believed to modulate platelet activity by downregulating inflammation and preventing thrombosis; however, there is no evidence to support that ASA actually prevents the development of CAA.[2][6] There has been some recent concern over ASA dosing in the acute phase of the disease due to hypoalbuminemia, slow gastric transition time, and high renal clearance, which causes toxic effects at lower doses.[3] It is also of note that children are at higher risk of developing Reye syndrome if they develop influenza or varicella infections while taking ASA, and children should receive the influenza vaccine and abstain from the varicella vaccine while taking ASA.[1] Clopidogrel or dipyridamole can be used as alternatives for ASA allergy, or temporarily during influenza or varicella illness to prevent Reye syndrome.
If the patient is still febrile 36 to 48 hours after the first IVIG dose, then the dose should be repeated once.[21] Ideally, IVIG should be started within 7-10 days of the onset of fever to prevent potential cardiac complications and can decrease CAA formation from 25% to 3-5%.[22] Some 15 to 20% of patients will fail IVIG treatment, with persistent fever for >36 hours after administration and require a second dose.[3][6] There has been evidence to suggest that pre-IVIG IgG levels correlate with both susceptibilities to therapy and cardiac outcomes.[23] Patients who have lower IgG levels before treatment have been shown to be more resistant to IVIG therapy and are at higher risk for developing CAAs.[1][5][24] Individuals at risk for IVIG resistance and development of CAA include those with gastrointestinal/abdominal involvement,[25] a higher percentage of bands, pro-BNP, CRP or alanine aminotransferase; or lower platelets or hemoglobin concentrations when compared to those who are susceptible.[3] There has been some concern about high dose IVIG (4g/kg vs. 2g/kg) causing hemolytic anemia, with patients who had complete KD, were refractory to IVIG, and non-O-blood groups being the most vulnerable.[26] Other alternatives to IVIG include infliximab at 5 mg/kg or cyclophosphamide.[27] Remember that IVIG can result in infusion reactions and pretreatment or access to resuscitative medications may be necessary. Also, remember that IVIG can result in ineffective vaccination, such that routine vaccines (such as MMR) may require delay.(A1)
Corticosteroids have been proposed as part of the initial therapy (in addition to IVIG and ASA) due to findings of decreased risk of developing cardiac abnormalities. However, research results have been inconsistent The RAISE trial published in 2012 reintroduced the idea of using extended doses of prednisolone (2mg/kg/day for 4 to 5 weeks) in patients with KD who were at high risk for being resistant to IVIG therapy.[28] There have been no findings suggestive that corticosteroids are harmful, and may be a therapeutic option at the discretion of the treating physician. Other studies suggest corticosteroids reduce the occurrence of CAAs, duration of fever, duration of hospitalization, and time taken to normalization of inflammatory markers.[1][3][21][29]and may be beneficial in cases refractory to initial IVIG therapy.[2][29] Recommended doses are 2 mg/kg of prednisone or its equivalent for 3 days to 2 weeks, depending on the individual's response.[3]
Some have proposed statins for KD due to their immunomodulating properties; however, there is not enough research at this time to suggest their use; the theory is that their ability to target endothelial dysfunction would make them of use during the acute phase to help prevent and/or modify vessel changes.[30] Angiotensin receptor blockers (ARBs) have also found use once a CAA was detected, and were shown to prevent stenosis due to hyper-proliferation.[4] Besides, there has been some evidence that ARBs and statins have an additive effect on one another to prevent atherosclerotic disease.[4] Plasma exchange therapy has demonstrated effectiveness in patients who are refractory to IVIG in reducing the incidence of CAAs; however, this has only been tested in small case series and uncontrolled clinical trials and is not a current recommendation.[1]
Patients who develop severe perfusion deficits from an aneurysm may require coronary artery bypass graft surgery and should be a consideration in children who have reversible ischemia or patients that have recurrent MIs.[1] Percutaneous intervention commonly fails due to the severity of disease and significant narrowing of the arteries, putting the patient at higher risk for new aneurysm formation or perforation.[30]
Long-term management starts after the acute illness has passed, typically 5-6 weeks after the initial onset of the fever; this is typically when coronary artery involvement has reached its peak in severity. The frequency of follow-up, medication, and repeat images are on a patient by patient basis and their disease severity.[2] However, both the AHA and Japanese Circulation Society (JCS) recommend routine cardiac stress testing for patients with medium-large CAAs as well as non-invasive imaging.[30] There is no consensus on the duration for ASA treatment, or whether there is any benefit of the patient being on dual anti-coagulant therapy after the acute phase of the disease. Low-molecular-weight-heparin or warfarin (with a goal of INR 2 to 2.5) is recommended in patients who have large CAAs, as well as an anti-platelet agent (clopidogrel or dipyridamole) to prevent thrombus formation due to decreased flow and the damaged epithelium.[1][4] Platelet activity remains elevated from 3 months to one year after disease onset, and patients should be on some type of low-dose anti-platelet therapy for 3 months.[4] The overall goal of long-term therapy is to prevent myocardial ischemia and infarction. Complete resolution of coronary artery lesions has been observed in over 50% of patients 1 to 2 years from the onset of disease and seems to be dependent on the size of the initial lesion.[1]
Differential Diagnosis
Various infections can mimic Kawasaki Disease including:
- Preseptal cellulitis
- Peritonsillar abscess
- Retropharyngeal Abscess
- Cervical lymphadenitis
- Group A streptococcal infection
- Adenovirus, Enterovirus, Parvovirus B19
- Measles
- Mononucleosis (Epstein-Barr virus)
- Scarlet Fever
- Rheumatic fever
- Toxic Shock Syndrome
- Meningitis
- Rocky Mountain Spotted Fever
- Staphylococcal scalded skin syndrome (SSSS)
- Toxic epidermal necrolysis (TEN)
- Lyme disease
- Leptospirosis
Both KD and adenovirus present with conjunctival injection, however, the important differentiation is that adenovirus causes conjunctival exudates and KD does not.[3] The differentiation of KD from lymphadenitis by observing whether the lymphadenopathy is bilateral or unilateral; Kawasaki typically presents unilaterally in over half of cases.[1][3] KD can cause retropharyngeal edema which may present concerning for possible retropharyngeal abscess (RPA). However, true RPA will have clinical symptoms and abnormal imaging, whereas KD will not.[3] Toxic shock syndrome and scarlet fever lack the ocular and joint involvement that KD has.[6]
Kawasaki disease' presentation also overlaps with other immunologic reactions such as multiple drug hypersensitivity reactions, juvenile idiopathic arthritis, infantile polyarteritis nodosa, and systemic lupus erythematosus. These can be differentiated from KD by the absence of classic clinical criteria and by chronicity and the number of joints affected.[6][12]
Pertinent Studies and Ongoing Trials
There are several clinical trials regarding the treatment of Kawasaki disease including the use of infliximab, etanercept, and anakinra. Infliximab has been found to decrease inflammatory markers (TNF) and downregulate macrophages in patients with IVIG resistant KD, and patients had a quicker resolution of fever but similar cardiac outcomes.[2][3][7] There has also been an investigative trial comparing using IVIG alone versus IVIG with infliximab for the initial treatment of children with KD. Children who received the combination treatment were found to be less refractory to treatment, had shorter fever durations and hospital stays, and had less coronary artery dilation.[26] Also, their lab abnormalities returned to normal values in a shorter duration. However, there was no difference in the actual occurrence of coronary aneurysms between the two groups.[26] Plasma exchange therapy has also been investigated as a potential therapy for patients refractory to IVIG treatment and has shown promise by demonstrating an improvement in and/or resolution of cardiac abnormalities during follow-up.[2] Abciximab has also been investigated for its use in KD patients. Its initial purpose was to target thrombi and vascular remodeling in adults who were suffering from coronary artery disease. However, recent studies have demonstrated that patients treated with abciximab have a quicker and greater reduction in their CAAs when compared to standard treatment of IVIG and ASA.[31][32] Cyclosporine A has also demonstrated promise for patients who are refractory to initial and subsequent IVIG treatments.[33] However, cyclosporine A has shown to cause arrhythmias and does not appear to influence endovascular inflammation and remodeling.[34]
Prognosis
Prognosis depends on the severity of cardiac disease. Additionally, the prognosis is better for children diagnosed between age 6 months to 9 years than those younger or older, perhaps because they are diagnosed earlier after presenting with classic findings.[35] Recurrence is uncommon but is most likely to occur in younger children who had cardiac complications from the disease during the initial episode. All KD related deaths are essentially the result of cardiac complications and typically occur 15-45 days from the onset of fever.[1]
Complications
The disease can lead to aneurysm formation, heart failure, MI, myocarditis, valvulitis, pericarditis with pericardial effusion, and rupture of the coronary arteries leading to hemopericardium and sudden death.[30] About 9% of patients will experience acute phase cardiac complications while about 3% will experience cardiac sequelae.[4] Patients with Kawasaki disease frequently have MIs while sleeping or at rest, which suggests coronary vasospasm etiology.[4] Myocardial inflammation has been seen in 50 to 70% of patients during the acute phase of the disease and has raised concern over the long-term effects that KD has on cardiac function.[1] There is documentation of mitral regurgitation and aortic regurgitation in a small number of cases in children with KD.[1] About 15-25% of untreated children will go on to develop CAAs.[1] Specific factors lead to an increased risk of coronary artery aneurysms including [1][4][5][10][11][12]:
- Fever >8 days (most important risk factor)
- Recurrence of fever after a period of being afebrile for 48 hours
- Male (3 times more likely to develop giant aneurysms)
- Cardiomegaly
- <1 year old of age
- Asian or Pacific Islander descent
- Hispanic ethnicity
- Lower IgG levels
- Increased pro-BNP levels
- Increased TNF-alpha levels
- Thrombocytopenia at initial presentation
- Incomplete Kawasaki Disease Diagnosis
Aneurysms are defined as vessel diameter >3mm in children younger than 5 years old and >4mm in children older than 5 years.[1] Cardiac complications later on in adulthood have been shown to be more common in those patients who had a CAA >6.0mm.[30] Giant aneurysms (>8.0mm) are likely to occlude from a thrombus within in the first year causing an MI.[19]
In those who aneurysms resolve, there will be significant thickening of the vessel leading to an increased risk of premature coronary atherosclerotic disease.[18] In a study done on adults who suffered from an MI later in life after having had a coronary artery aneurysm in childhood due to KD, there was no evidence of remaining aneurysm.[36] However, one cannot assume that just because an aneurysm resolves, that does not mean the risk of cardiac complications later in life also dissipates.[30]
Pearls and Other Issues
If KD is on your differential, order the echocardiogram; missing a diagnosis or diagnosing the disease late can be catastrophic.
If considering incomplete KD, order ESR, CRP, CBC, serum albumin and alanine aminotransferase and urinalysis.
Missed diagnosis is often due to vague initial presentation, typically with fever and enlarged unilateral lymph nodes. The following rash and mucosal changes may be mistakenly blamed on a reaction to the antibiotics for presumed bacterial infection.[1] Pyuria can be mistaken for a UTI, and pleocytosis of the CSF can be mistaken for viral meningitis.[1] Many KD patients with gastroenteritis or viral respiratory symptoms and a positive viral PCR test may be misleading.
Children with Kawasaki disease should get an echocardiogram at the time of diagnosis, two weeks after symptom onset, and again at eight weeks after the diagnosis. AHA recommends repeating an echocardiogram 10 to 14 days after the initial one and 4 to 6 weeks after all laboratory data have normalized.
The goal of treatment is the prevention of cardiac complications. Therefore, all patients diagnosed with Kawasaki disease should receive IVIG and aspirin.
Many children get lost to long-term follow-up as an adult leading to severe cardiac complications.
Enhancing Healthcare Team Outcomes
Kawasaki disease is a rare disorder, but because of its serious effects on the coronaries, the condition is best managed by an interprofessional team. Once a child is suspected of having Kawasaki disease, the patient should be referred promptly to a cardiologist.
A cardiologist should be consulted to determine the initial and schedule of following echocardiogram studies, anticoagulation management, to determine if coronary artery angiography is necessary and for long-term follow-up and monitoring. Other specialists to consider, include an infectious disease specialist, rheumatologist, or dermatologist. Following treatment, the short-term prognosis is good, but the long-term prognosis remains unknown primarily because many children are lost to followup.
Media
(Click Image to Enlarge)
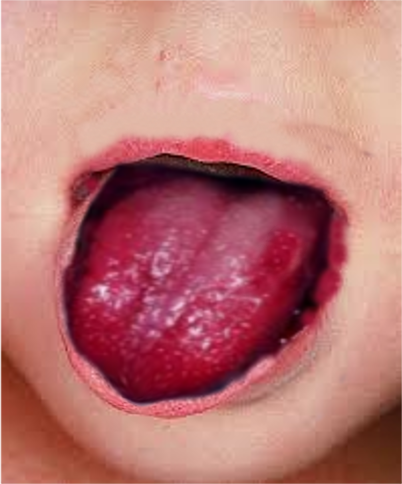
Strawberry tongue KD Image courtesy S Bhimji MD
References
McCrindle BW, Rowley AH, Newburger JW, Burns JC, Bolger AF, Gewitz M, Baker AL, Jackson MA, Takahashi M, Shah PB, Kobayashi T, Wu MH, Saji TT, Pahl E, American Heart Association Rheumatic Fever, Endocarditis, and Kawasaki Disease Committee of the Council on Cardiovascular Disease in the Young; Council on Cardiovascular and Stroke Nursing; Council on Cardiovascular Surgery and Anesthesia; and Council on Epidemiology and Prevention. Diagnosis, Treatment, and Long-Term Management of Kawasaki Disease: A Scientific Statement for Health Professionals From the American Heart Association. Circulation. 2017 Apr 25:135(17):e927-e999. doi: 10.1161/CIR.0000000000000484. Epub 2017 Mar 29 [PubMed PMID: 28356445]
Saguil A, Fargo M, Grogan S. Diagnosis and management of kawasaki disease. American family physician. 2015 Mar 15:91(6):365-71 [PubMed PMID: 25822554]
Vervoort D, Donné M, Van Gysel D. Pitfalls in the diagnosis and management of Kawasaki disease: An update for the pediatric dermatologist. Pediatric dermatology. 2018 Nov:35(6):743-747. doi: 10.1111/pde.13620. Epub 2018 Oct 18 [PubMed PMID: 30338568]
Guidelines for diagnosis and management of cardiovascular sequelae in Kawasaki disease (JCS 2013). Digest version. Circulation journal : official journal of the Japanese Circulation Society. 2014 [PubMed PMID: 25241888]
Turnier JL, Anderson MS, Heizer HR, Jone PN, Glodé MP, Dominguez SR. Concurrent Respiratory Viruses and Kawasaki Disease. Pediatrics. 2015 Sep:136(3):e609-14. doi: 10.1542/peds.2015-0950. Epub 2015 Aug 24 [PubMed PMID: 26304824]
Sundel RP. Kawasaki disease. Rheumatic diseases clinics of North America. 2015:41(1):63-73, viii. doi: 10.1016/j.rdc.2014.09.010. Epub [PubMed PMID: 25399940]
Matsubara T. Infliximab for the treatment of Kawasaki disease. Pediatrics international : official journal of the Japan Pediatric Society. 2018 Sep:60(9):775. doi: 10.1111/ped.13663. Epub [PubMed PMID: 30255976]
Kim DS. Kawasaki disease. Yonsei medical journal. 2006 Dec 31:47(6):759-72 [PubMed PMID: 17191303]
Takahashi K, Oharaseki T, Yokouchi Y. Pathogenesis of Kawasaki disease. Clinical and experimental immunology. 2011 May:164 Suppl 1(Suppl 1):20-2. doi: 10.1111/j.1365-2249.2011.04361.x. Epub [PubMed PMID: 21447126]
Rowley AH, Baker SC, Shulman ST, Garcia FL, Fox LM, Kos IM, Crawford SE, Russo PA, Hammadeh R, Takahashi K, Orenstein JM. RNA-containing cytoplasmic inclusion bodies in ciliated bronchial epithelium months to years after acute Kawasaki disease. PloS one. 2008 Feb 13:3(2):e1582. doi: 10.1371/journal.pone.0001582. Epub 2008 Feb 13 [PubMed PMID: 18270572]
Level 2 (mid-level) evidenceLang B. Recognizing Kawasaki disease. Paediatrics & child health. 2001 Nov:6(9):638-43 [PubMed PMID: 20084135]
Marchesi A, Tarissi de Jacobis I, Rigante D, Rimini A, Malorni W, Corsello G, Bossi G, Buonuomo S, Cardinale F, Cortis E, De Benedetti F, De Zorzi A, Duse M, Del Principe D, Dellepiane RM, D'Isanto L, El Hachem M, Esposito S, Falcini F, Giordano U, Maggio MC, Mannarino S, Marseglia G, Martino S, Marucci G, Massaro R, Pescosolido C, Pietraforte D, Pietrogrande MC, Salice P, Secinaro A, Straface E, Villani A. Kawasaki disease: guidelines of the Italian Society of Pediatrics, part I - definition, epidemiology, etiopathogenesis, clinical expression and management of the acute phase. Italian journal of pediatrics. 2018 Aug 30:44(1):102. doi: 10.1186/s13052-018-0536-3. Epub 2018 Aug 30 [PubMed PMID: 30157897]
Rowley AH, Shulman ST. Kawasaki syndrome. Clinical microbiology reviews. 1998 Jul:11(3):405-14 [PubMed PMID: 9665974]
Jindal AK, Indla RT, Pilania RK, Singh S. Non-Exudative Conjunctival Injection With Limbal Sparing: A Pathognomonic Clinical Sign of Kawasaki Disease. Journal of clinical rheumatology : practical reports on rheumatic & musculoskeletal diseases. 2020 Apr:26(3):e59-e60. doi: 10.1097/RHU.0000000000000883. Epub [PubMed PMID: 30247222]
Beken B, Unal S, Cetin M, Gümrük F. The relationship between hematological findings and coronary artery aneurysm in kawasaki disease. Turkish journal of haematology : official journal of Turkish Society of Haematology. 2014 Jun:31(2):199-200. doi: 10.4274/tjh.2013.0241. Epub 2014 Jun 10 [PubMed PMID: 25035683]
Suddleson EA, Reid B, Woolley MM, Takahashi M. Hydrops of the gallbladder associated with Kawasaki syndrome. Journal of pediatric surgery. 1987 Oct:22(10):956-9 [PubMed PMID: 3316594]
Sun Q, Zhang J, Yang Y. Gallbladder Hydrops Associated With Kawasaki Disease: A Case Report and Literature Review. Clinical pediatrics. 2018 Mar:57(3):341-343. doi: 10.1177/0009922817696468. Epub 2017 Mar 1 [PubMed PMID: 28952362]
Level 3 (low-level) evidenceReddy S, Forbes T, Chintala K. Cardiovascular involvement in Kawaski Disease. Images in paediatric cardiology. 2005 Apr:7(2):1-9 [PubMed PMID: 22368648]
Level 3 (low-level) evidenceTsuda E, Singhal M. Role of imaging studies in Kawasaki disease. International journal of rheumatic diseases. 2018 Jan:21(1):56-63. doi: 10.1111/1756-185X.13210. Epub 2017 Nov 8 [PubMed PMID: 29115035]
Kahn AM, Budoff MJ, Daniels LB, Jimenez-Fernandez S, Cox AS, Gordon JB, Burns JC. Calcium scoring in patients with a history of Kawasaki disease. JACC. Cardiovascular imaging. 2012 Mar:5(3):264-72. doi: 10.1016/j.jcmg.2011.12.010. Epub [PubMed PMID: 22421171]
Level 2 (mid-level) evidenceBurns JC, Capparelli EV, Brown JA, Newburger JW, Glode MP. Intravenous gamma-globulin treatment and retreatment in Kawasaki disease. US/Canadian Kawasaki Syndrome Study Group. The Pediatric infectious disease journal. 1998 Dec:17(12):1144-8 [PubMed PMID: 9877364]
Level 2 (mid-level) evidenceDurongpisitkul K, Gururaj VJ, Park JM, Martin CF. The prevention of coronary artery aneurysm in Kawasaki disease: a meta-analysis on the efficacy of aspirin and immunoglobulin treatment. Pediatrics. 1995 Dec:96(6):1057-61 [PubMed PMID: 7491221]
Level 1 (high-level) evidenceKim HK, Oh J, Hong YM, Sohn S. Parameters to guide retreatment after initial intravenous immunoglobulin therapy in kawasaki disease. Korean circulation journal. 2011 Jul:41(7):379-84. doi: 10.4070/kcj.2011.41.7.379. Epub 2011 Jul 30 [PubMed PMID: 21860639]
Yamazaki-Nakashimada MA, Gámez-González LB, Murata C, Honda T, Yasukawa K, Hamada H. IgG levels in Kawasaki disease and its association with clinical outcomes. Clinical rheumatology. 2019 Mar:38(3):749-754. doi: 10.1007/s10067-018-4339-0. Epub 2018 Oct 20 [PubMed PMID: 30343342]
Level 2 (mid-level) evidenceFabi M, Corinaldesi E, Pierantoni L, Mazzoni E, Landini C, Bigucci B, Ancora G, Malaigia L, Bodnar T, Di Fazzio G, Lami F, Valletta E, Cicero C, Biasucci G, Iughetti L, Marchetti F, Sogno Valin P, Amarri S, Brusa S, Sprocati M, Maggiore G, Dormi A, Lanzoni P, Donti A, Lanari M. Gastrointestinal presentation of Kawasaki disease: A red flag for severe disease? PloS one. 2018:13(9):e0202658. doi: 10.1371/journal.pone.0202658. Epub 2018 Sep 4 [PubMed PMID: 30180185]
Nolan BE, Wang Y, Pary PP, Luban NLC, Wong ECC, Ronis T. High-dose intravenous immunoglobulin is strongly associated with hemolytic anemia in patients with Kawasaki disease. Transfusion. 2018 Nov:58(11):2564-2571. doi: 10.1111/trf.14879. Epub 2018 Sep 28 [PubMed PMID: 30265742]
Newburger JW. Kawasaki disease: Medical therapies. Congenital heart disease. 2017 Sep:12(5):641-643. doi: 10.1111/chd.12502. Epub 2017 Jun 5 [PubMed PMID: 28580631]
Burns JC, Hoshino S, Kobayashi T. Kawasaki disease: an essential comparison of coronary artery aneurysm criteria. The Lancet. Child & adolescent health. 2018 Dec:2(12):840-841. doi: 10.1016/S2352-4642(18)30334-1. Epub 2018 Oct 16 [PubMed PMID: 30337184]
Miyata K, Kaneko T, Morikawa Y, Sakakibara H, Matsushima T, Misawa M, Takahashi T, Nakazawa M, Tamame T, Tsuchihashi T, Yamashita Y, Obonai T, Chiga M, Hori N, Komiyama O, Yamagishi H, Miura M, Post RAISE group. Efficacy and safety of intravenous immunoglobulin plus prednisolone therapy in patients with Kawasaki disease (Post RAISE): a multicentre, prospective cohort study. The Lancet. Child & adolescent health. 2018 Dec:2(12):855-862. doi: 10.1016/S2352-4642(18)30293-1. Epub 2018 Oct 16 [PubMed PMID: 30337183]
Denby KJ, Clark DE, Markham LW. Management of Kawasaki disease in adults. Heart (British Cardiac Society). 2017 Nov:103(22):1760-1769. doi: 10.1136/heartjnl-2017-311774. Epub 2017 Jul 27 [PubMed PMID: 28751537]
Williams RV, Wilke VM, Tani LY, Minich LL. Does Abciximab enhance regression of coronary aneurysms resulting from Kawasaki disease? Pediatrics. 2002 Jan:109(1):E4 [PubMed PMID: 11773572]
McCandless RT,Minich LL,Tani LY,Williams RV, Does abciximab promote coronary artery remodeling in patients with Kawasaki disease? The American journal of cardiology. 2010 Jun 1 [PubMed PMID: 20494673]
Level 2 (mid-level) evidenceSuzuki H, Terai M, Hamada H, Honda T, Suenaga T, Takeuchi T, Yoshikawa N, Shibuta S, Miyawaki M, Oishi K, Yamaga H, Aoyagi N, Iwahashi S, Miyashita R, Onouchi Y, Sasago K, Suzuki Y, Hata A. Cyclosporin A treatment for Kawasaki disease refractory to initial and additional intravenous immunoglobulin. The Pediatric infectious disease journal. 2011 Oct:30(10):871-6. doi: 10.1097/INF.0b013e318220c3cf. Epub [PubMed PMID: 21587094]
Level 3 (low-level) evidenceKuijpers TW, Biezeveld M, Achterhuis A, Kuipers I, Lam J, Hack CE, Becker AE, van der Wal AC. Longstanding obliterative panarteritis in Kawasaki disease: lack of cyclosporin A effect. Pediatrics. 2003 Oct:112(4):986-92 [PubMed PMID: 14523200]
Level 3 (low-level) evidenceGupta-Malhotra M, Gruber D, Abraham SS, Roman MJ, Zabriskie JB, Hudgins LC, Flynn PA, Levine DM, Okorie U, Baday A, Schiller MS, Maturi J, Meehan D, Dyme J, Parker TS, Wittkowski KM, Gersony WM, Cooper RS. Atherosclerosis in survivors of Kawasaki disease. The Journal of pediatrics. 2009 Oct:155(4):572-7. doi: 10.1016/j.jpeds.2009.04.054. Epub [PubMed PMID: 19595365]
Level 2 (mid-level) evidenceGordon JB, Kahn AM, Burns JC. When children with Kawasaki disease grow up: Myocardial and vascular complications in adulthood. Journal of the American College of Cardiology. 2009 Nov 17:54(21):1911-20. doi: 10.1016/j.jacc.2009.04.102. Epub [PubMed PMID: 19909870]