Introduction
Neurons are electrically excitable, reacting to input via the production of electrical impulses, propagated as action potentials throughout the cell and its axon. These action potentials are generated and propagated by changes to the cationic gradient (mainly sodium and potassium) across their plasma membranes. These action potentials finally reach the axonal terminal and cause depolarization of neighboring cells through synapses. This action is the way these cells can interact with each other, i.e., at synapses via synaptic transmission. Normally, the cell’s interior is negative, compared to its outside. This state is the resting membrane potential of about -60mV. A neuronal action potential gets generated when the negative inside potential reaches the threshold (less negative). This change in membrane potential will open voltage-gated cationic channel (sodium channel) resulting in the process of depolarization and generation of the neuronal action potential. Neuronal action potentials are vital for propagation of impulses along any nerve fiber even at a distance. They also are crucial for communication among neurons through synapses. Disruption of this mechanism can have drastic effects resulting in lack of impulse generation and conduction, illustrated by various neurotoxins and demyelinating disorders.[1][2]
Structure and Function
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Structure and Function
The neuron’s membrane potential gets generated via a difference in the concentration of charged ions. The lipid bilayer of the neuronal cell membrane acts as a capacitor, the transmembrane channels as resistors. This resting (steady-state) potential is critical for the neuron’s physiological state, maintained by an unequal distribution of ions across the cellular membrane and established by ATP-dependent pumps--most notably, sodium-potassium antiporters. These exchangers are responsible for pumping sodium out of the cells into the extracellular space, potassium into the intracellular compartment. When opened, various channels allow permeable ions to flow down their electrochemical gradients, thereby altering the membrane potential. The gating of these channels is by second messengers, neurotransmitters, or voltage changes. Voltage-gated cationic channels are the main channels used in the generation and propagation of neuronal action potential.
There are 100 billion neurons in the human brain, and there are a quadrillion synapses in the human brain. Any neuron will have on average of 1000 synapses which influence the electrical potential of the membrane. When the resting membrane potential (-60mV) becomes less negative, it depolarizes. When it is more negative, it hyperpolarizes. Upon collating the various movements of ions, particularly the entering of sodium, the cell may have sufficient signals to reach the threshold potential and achieves this threshold by sufficient positively charged ions entering the cell, i.e., terminating the polarity in what is called depolarization. At normal body temperature, the equilibrium potential for sodium is +55 mV, -103 mV for potassium. There are three stages in the generation of the action potential: (1) depolarization, changing the membrane’s potential from -60 mV to +40 mV primarily caused by sodium influx; (2) repolarization, a return to the membrane’s resting potential, primarily caused by potassium efflux; and (3) after-hyperpolarization, a recovery from a slight overshoot of the repolarization.[3] (see table below) As mentioned, stage 1 is guided by an increased membrane permeability to sodium. Accordingly, the removal of extracellular sodium, or inactivation of sodium channels, prevents the generation of action potentials.[4] Immediately after an action potential generates, the neuron cannot immediately generate another action potential; this is the absolute refractory period. At this moment, the sodium channels are inactivated and remain closed, whereas the potassium channels are still open. This state is followed by the relative refractory period when the neuron may only generate an action potential with a much higher threshold. Thie opens when some of the sodium channels are ready to be opened, and many are still inactivated, whereas some potassium channels are still open as well. The duration of the refractory periods will determine how fast an action potential may be generated and propagated. The propagation of the action potential continues until termination at a synapse, where it can either cause the release of neurotransmitters or conduction of ionic currents. The latter occurs at electrical synapses, whereby presynaptic and postsynaptic cells connect and avoid the use of neurotransmitters.[5] Neurotransmitters are the norm, however, and get released at chemical synapses and neuromuscular junctions.[6]
Local currents created by depolarization along a portion of the neuronal membrane, if sufficiently strong, can depolarize neighboring segments of the membrane to the threshold, thereby propagating the action threshold down the membrane and along the neuron’s axon. The determining factor in the speed of this propagation is primarily the extent to which the initial local currents first spread before creating further depolarizations. Factors influencing this speed include the membrane’s electrical resistance and internal contents of the axon. Wider axons have lower internal resistance, and having more voltage-gated sodium channels in the membrane decreases membrane resistance as well. Higher internal resistance and lower membrane resistance contribute to slower action potential propagations. Because the body does not have enough space, instead of making large axons, the nervous system, to maximize propagation velocity, employs glial cells, specifically oligodendrocytes and Schwann cells, to wrap themselves around axons, creating myelin sheaths. These sheaths contribute to greater membrane resistance, patching up areas where channels would otherwise leak. Still, the action potential can only propagate so far before requiring more sodium channels to perpetuate the potential, creating gaps in the myelin sheath called nodes of Ranvier. These nodes have high concentrations of those channels to restart the action potential along the axon, termed saltatory conduction.[1]
Neuron Action Potential - see the table in media below.
Clinical Significance
The rapid depolarization or the upstroke of the neuronal action potential occurs as a result of the opening of the voltage-gated sodium channels. These channels are large transmembrane proteins with different subunits encoded by ten mammalian genes. Problems with these channels are collectively called channelopathies. The channelopathies may affect any excitable tissues, including neurons, skeletal, and cardiac muscles resulting in multiple different diseases. The neurological channelopathies present more commonly in different muscle diseases and the brain. Paramyotonia congenita results from mutations in the gene coding for the alpha-1 subunit of the sodium channel. Sodium channelopathies in the brain result in various forms of refractory epilepsy disorders.
There is a variety of neurotoxins that can block the action potential. One such deadly toxin is tetrodotoxin (TTX), which inhibits sodium channels.[7] The naturally occurring toxin is normally ingested orally from pufferfish, a part of Japanese cuisine, and its incidence has spread beyond Southeast Asia to the Pacific and Mediterranean, as well as finding this toxin in many other species. By binding to sodium channels and inactivating them, tissues affected are rendered immobile and insensitive. The onset/severity of symptoms arising from TTX correlates on how much an individual consumes, and patients may first present with paraesthesias of the tongue/lips. This presentation is associated with or followed by headache/vomiting that may become muscle weakness and ataxia. Other symptoms include diarrhea, dizziness, and loss of reflexes. Death can occur from respiratory and/or heart failure. Of some clinical significance, however, TTX has some analgesic activity that has been the topic of study in treating pain, and a low dose may reduce heroin craving. Unfortunately, TTX has no cure and is often fatal, with observation and supportive care being the only treatment. Respiratory support comes in the form of endotracheal intubation or mechanical ventilation to support breathing. Early stages of poisoning can be treated with activated charcoal to adsorb the toxin before gastric absorption and with gastric lavage to reduce symptom severity.[8]
Ciguatoxin is a potent sodium channel blocker that causes a rapid onset of numbness, paraesthesia, dysaesthesia, and muscle paralysis. Ciguatoxins (CTX) are marine neurotoxins that are produced by the dinoflagellates. CTX works by blocking the voltage-gated sodium channels. Humans are exposed to CTX by ingestion of carnivore coral reef fishes, including grouper, red snapper, and barracuda, which feed on fish that have consumed the dinoflagellates.
Saxitoxin and its derivatives are known as paralytic shellfish toxins (PSTs). Found in marine and freshwater environments among dinoflagellates, PSTs act similarly to TTX and CTX, i.e., binding to voltage-gated sodium channels and blocking the movement of nerve impulses, as well as some degree of targeting the potassium and calcium channels. As such, just like TTX, sodium cannot enter through the inactivated sodium channels, preventing membrane depolarization. Because of the similarity of its mechanism of action to TTX, PSTs share similar consequences. Severe exposure can cause severe hypotension and general paralysis, and death can occur from respiratory failure/hypotension.[9][10]
To illustrate the importance of myelin for saltatory conduction, different demyelinating diseases that destroy myelin can have varying degrees of severity because they reduce the conduction velocity of action potentials.[11] Multiple sclerosis (MS) destroys oligodendrocytes, which help to maintain the fatty layer of the myelin sheath, preventing the effective carriage of electrical signals. Eventually, this causes total loss of myelin and breakdown of neuronal axons.[12] MS commonly presents in white young adult females and can result in a plethora of signs/symptoms, physical, mental, and psychiatric, e.g., diplopia, blindness, muscle weakness, speech problems, tremors, incontinence, and vertigo. Diagnosis can be aided with testing for oligoclonal bands of IgG in cerebrospinal fluid on electrophoresis, found in many MS patients.[13][14][15][16]
Media
(Click Image to Enlarge)
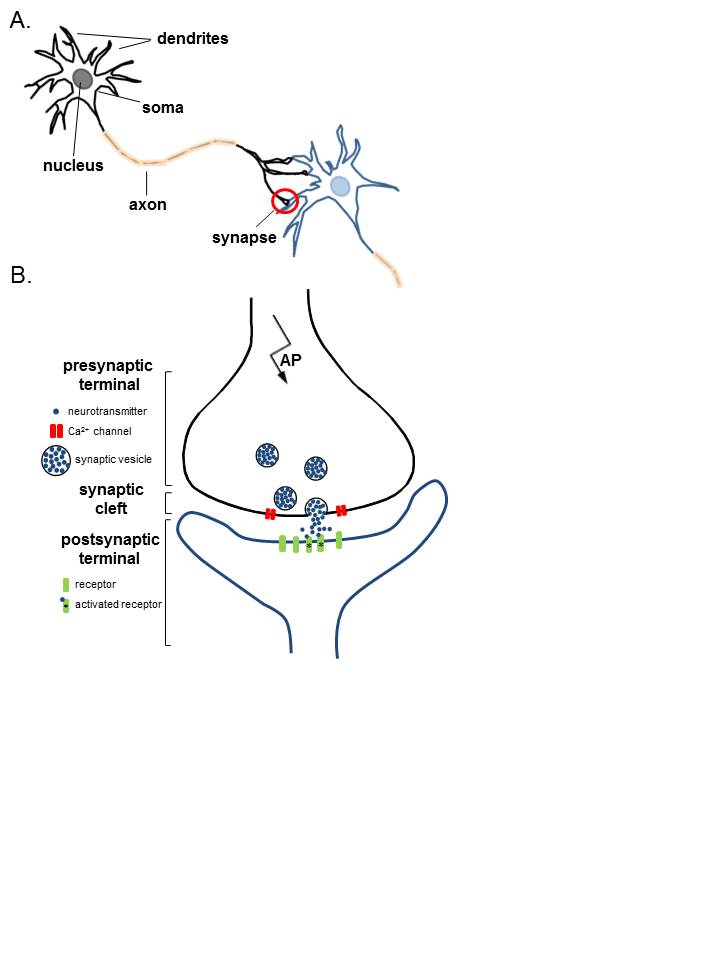
Anatomy of Neurons. A. Two connected neurons. Neurons have a soma that contains a nucleus, an axon, and a dendritic tree. A single synapse (red circle) is formed at the point where an axon's neuron (black) connects to another neuron's dendrite, soma, or axon (blue). B. A magnified view of a single synapse. On the arrival of an action potential at the presynaptic terminal, calcium triggers the release of neurotransmitters from the synaptic vesicles into the synaptic cleft. Neurotransmitters diffuse across the synaptic cleft to activate postsynaptic receptors.
Contributed Image by Karin Aubrey
(Click Image to Enlarge)
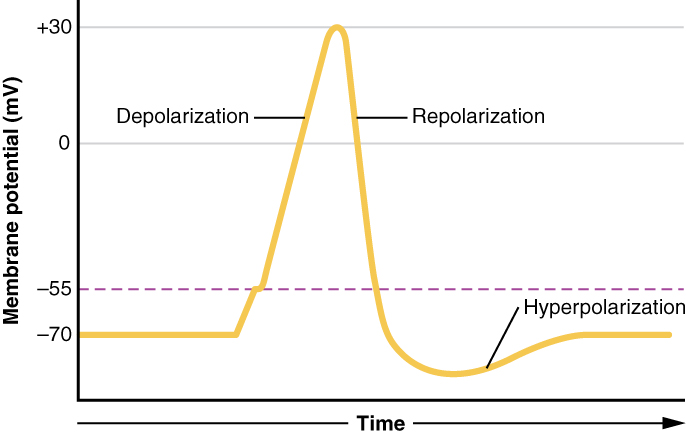
A neuronal action potential. The dashed line represents the threshold voltage. Used with permission from OpenStax under the Creative Commons Attribution 4.0 International license.
(Click Image to Enlarge)
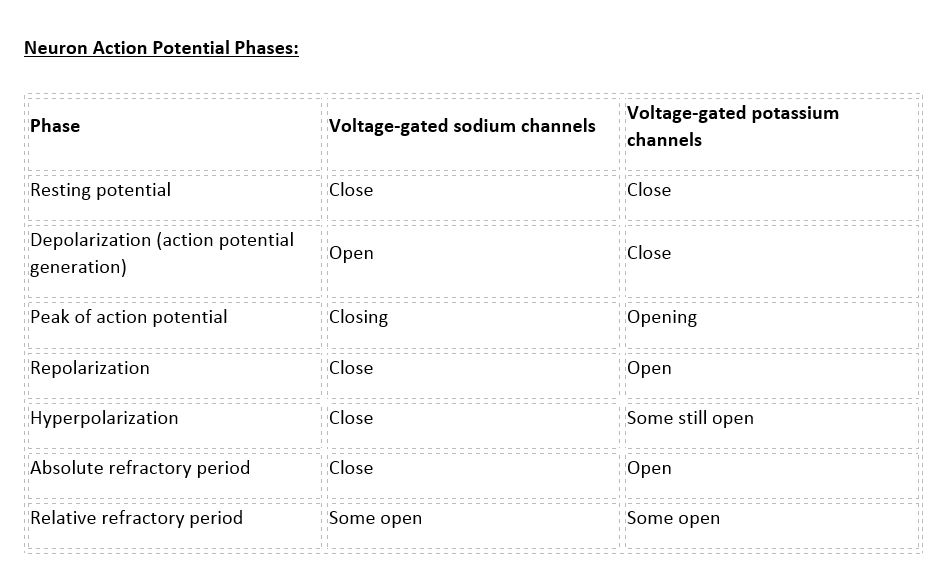
Neuron action potential - ionic movements Contributed by Forshing Lui MD
References
Hodgkin AL, Huxley AF. A quantitative description of membrane current and its application to conduction and excitation in nerve. 1952. Bulletin of mathematical biology. 1990:52(1-2):25-71; discussion 5-23 [PubMed PMID: 2185861]
Level 3 (low-level) evidenceBarnett MW, Larkman PM. The action potential. Practical neurology. 2007 Jun:7(3):192-7 [PubMed PMID: 17515599]
Grider MH, Jessu R, Kabir R. Physiology, Action Potential. StatPearls. 2023 Jan:(): [PubMed PMID: 30844170]
Rutecki PA. Neuronal excitability: voltage-dependent currents and synaptic transmission. Journal of clinical neurophysiology : official publication of the American Electroencephalographic Society. 1992 Apr:9(2):195-211 [PubMed PMID: 1375602]
Level 3 (low-level) evidenceZoidl G, Dermietzel R. On the search for the electrical synapse: a glimpse at the future. Cell and tissue research. 2002 Nov:310(2):137-42 [PubMed PMID: 12397368]
Level 3 (low-level) evidenceSheffler ZM, Reddy V, Pillarisetty LS. Physiology, Neurotransmitters. StatPearls. 2023 Jan:(): [PubMed PMID: 30969716]
Jal S, Khora SS. An overview on the origin and production of tetrodotoxin, a potent neurotoxin. Journal of applied microbiology. 2015 Oct:119(4):907-16. doi: 10.1111/jam.12896. Epub 2015 Aug 13 [PubMed PMID: 26178523]
Level 3 (low-level) evidenceBane V, Lehane M, Dikshit M, O'Riordan A, Furey A. Tetrodotoxin: chemistry, toxicity, source, distribution and detection. Toxins. 2014 Feb 21:6(2):693-755. doi: 10.3390/toxins6020693. Epub 2014 Feb 21 [PubMed PMID: 24566728]
Level 3 (low-level) evidenceDiehl F, Ramos PB, Dos Santos JM, Barros DM, Yunes JS. Behavioral alterations induced by repeated saxitoxin exposure in drinking water. The journal of venomous animals and toxins including tropical diseases. 2016:22():18. doi: 10.1186/s40409-016-0072-9. Epub 2016 May 17 [PubMed PMID: 27190499]
Level 3 (low-level) evidenceCusick KD, Sayler GS. An overview on the marine neurotoxin, saxitoxin: genetics, molecular targets, methods of detection and ecological functions. Marine drugs. 2013 Mar 27:11(4):991-1018. doi: 10.3390/md11040991. Epub 2013 Mar 27 [PubMed PMID: 23535394]
Level 3 (low-level) evidenceMiller RH, Mi S. Dissecting demyelination. Nature neuroscience. 2007 Nov:10(11):1351-4 [PubMed PMID: 17965654]
Level 3 (low-level) evidenceCompston A, Coles A. Multiple sclerosis. Lancet (London, England). 2002 Apr 6:359(9313):1221-31 [PubMed PMID: 11955556]
Compston A, Coles A. Multiple sclerosis. Lancet (London, England). 2008 Oct 25:372(9648):1502-17. doi: 10.1016/S0140-6736(08)61620-7. Epub [PubMed PMID: 18970977]
Milo R, Kahana E. Multiple sclerosis: geoepidemiology, genetics and the environment. Autoimmunity reviews. 2010 Mar:9(5):A387-94. doi: 10.1016/j.autrev.2009.11.010. Epub 2009 Nov 20 [PubMed PMID: 19932200]
Level 3 (low-level) evidenceKhan O, Williams MJ, Amezcua L, Javed A, Larsen KE, Smrtka JM. Multiple sclerosis in US minority populations: Clinical practice insights. Neurology. Clinical practice. 2015 Apr:5(2):132-142 [PubMed PMID: 26137421]
Link H, Huang YM. Oligoclonal bands in multiple sclerosis cerebrospinal fluid: an update on methodology and clinical usefulness. Journal of neuroimmunology. 2006 Nov:180(1-2):17-28 [PubMed PMID: 16945427]
Level 3 (low-level) evidence