Introduction
Nociception provides a means of neural feedback that allows the central nervous system (CNS) to detect and avoid noxious and potentially damaging stimuli in both active and passive settings.[1][2][3][4][5][6] The sensation of pain divides into four large types: acute pain, nociceptive pain, chronic pain, and neuropathic pain. This article will consider the categories of acute and nociceptive pain together. Acute noxious stimuli (e.g., heat, cold, mechanical force, or chemical stimulation) trigger nociceptors. Acute pain becomes inflammatory pain when the noxious stimulus persists long enough to allow nociceptive neurons to release their pro-inflammatory markers and sensitize or activate responsive cells in their local environment.[7][8] Nociceptive pain arises from tissues damaged by physical or chemical agents such as trauma, surgery, or chemical burns, while neuropathic pain arises from diseases or damage mediated directly to sensory nerves, such as diabetic neuropathy, shingles, or postherpetic neuralgia.[2][6][8] Differentiating acute and nociceptive pain from neuropathic pain aids in understanding the broader study of pain; however, neuropathic pain will not be evaluated further in this article.
Regarding active settings, stimulated nociceptive neurons convey high-threshold noxious stimuli to the CNS. The nociceptive signal may either get redirected immediately in a spinal reflex loop, producing a rapid and reflexive withdrawal or transported to the areas of the brain responsible for integrating the information with higher-ordered sensations such as pain.[2][3][4][6][7] In addition to spinal afferent transmission to the CNS, nociceptive neurons are also capable of responding to noxious stimuli by secreting chemical signals from their peripheral nerve endings.[1][2] Local actions on nearby neuronal and non-neural cells undergo mediation through the release of vesicles containing preformed pro-inflammatory cytokines and growth factors.[7][8]
Depending on the specific monomodal sensitivity of a previously inactive nociceptor, specific noxious stimuli are detected by expressed receptors that open their cation channels in response to activation. The open cation channels on the nociceptive neurons depolarize the nociceptor, inducing vesicle fusion and cytokine release. The cytokines are pro-inflammatory, and once released, they elicit and propagate a matched release of pro-inflammatory cytokines from local epithelial, endothelial, and lymphoid cells.[1] The responding cells may then migrate or otherwise disseminate their pro-inflammatory signals that go on to sensitize or activate surrounding nociceptors originally outside of the primary nociceptive field.[7]
The spread of nociceptor-induced inflammation occurring over an area greater than that of the original nociceptor(s) involved is referred to as neuroinflammation. The propagation from nociceptive neurons to the surrounding cells, which may in-turn sensitize nearby nociceptive neurons, is why neuroinflammation is considered to be a self-reinforcing phenomenon.[1][7] Not only do the released pro-inflammatory molecules activate local inflammatory cells, but they are also capable of directly activating other nociceptive nerve endings because almost all nociceptive nerve endings possess receptors for all of the pro-inflammatory markers they are capable of releasing.[7] The pro-inflammatory molecules released from a directly stimulated nociceptive neuron are capable of binding to and activating a local nociceptive neuron entirely unaffected by the original stimulus. As with direct activation, the pro-inflammatory molecules bind the receptors on nociceptive nerve endings and depolarize the cell. Depolarization induces mitogen and protein activated kinases that phosphorylate other transducer proteins, such as TRPV1. This will activate and reinforce the depolarization, which, if of sufficient amplitude, will recruit voltage-gated sodium channels and truly depolarize the nerve fiber.[7]
Nociceptive signals cease with the termination of the stimulus, dephosphorylation, and suppression of the receptor, or once the influx of calcium through the open membrane proteins induces the nociceptive nerve ending to collapse and become refractory to restimulation in either neuronal or secretory mechanisms.[1] The collapse of the nociceptor following stimulation supports the finding that noxious stimuli quickly adjust, and their conscious perception abates quickly once their peripheral activity ceases.[1]
There is also a mostly unexplored role of passive nociception. Passive nociception refers to the involvement of inactive nociceptors, by their presence and previous activations, in guiding conscious actions so that the individual performs them in a manner least likely to produce pain or injury. Inactive nociceptors may provide less-than conscious "nudges" that strongly encourage the avoidance of potentially injurious and hazardous exposures.[4][9] Baliki and Apkarian presented this explanation and differentiated the subconscious, unconscious, or preconscious processes through which nociception may guide behaviors from those more active processes that steer actions through the conscious and subjective experience of pain.[9] Their results demonstrate the effects that an absence of nociceptive input may have in three poignant studies; the general disregard for injury seen in patients with painless channelopathies, the self-destructive gait seen in patients rendered insensitive to pain due to leprosy, and the boney destruction of weight-bearing joints seen in patients with painless Charcot joints of tabes dorsalis.[9] They postulate that the act of walking with proper gait, sitting with proper posture, or standing and stretching at regular intervals during long sedentary periods spent studying, are unconsciously motivated by nociceptive signals.[9] These studies illustrate the various injuries that are possible in the absence of the protective nociceptive signals that protect the body from avoidable joint injuries, muscle spasms, or pressure ulcers that would otherwise be difficult to imagine in an unafflicted individual capable of perceiving and avoiding these injuries.[9]
Issues of Concern
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Issues of Concern
Nociceptive fibers differentiated into four distinct subtypes according to their physical characteristics and potential sensory stimuli transmitted:
Aalpha-fibers: function in proprioception, no thermal sensitivity, myelinated, a diameter of 13 to 20 um, conduction speeds of 80 to 120 m/s, carry information regarding the position of limbs, muscles, and joint capsules.[4][7]
Abeta-fibers: function in mechanoreception (1 to 1.5 mN force threshold), no thermal sensitivity, myelinated, a diameter of 6 to 12 um, conduction speeds of 35 to 90 m/s, carry touch and pressure information from the skin.[4][7]
Adelta-fibers: function in nociception (5 mN force threshold), thermally sensitive, myelinated, a diameter of 1 to 5 um, conduction speeds of 5 to 40 m/s, carry heat, cold, and mechanothermal information from the skin, alternatively; subpopulations without thermal sensitivity instead carry information regarding touch from the skin.[5][7][10]
C-fibers: function in nociception, thermally sensitive, unmyelinated, a diameter of 0.02 to 1.5 um, conduction speeds of 0.5 to 2 m/s, carry polymodal information regarding mechanical, thermal, and/or chemical nociception; subpopulations include C-mechanonociceptors, C-mechanoheatnociceptors, C-mechanocoldnociceptors, C-heat nociceptors, and silent fibers only become heat-sensitive following sensitization.[5][7][10]
In summary, all A-fibers are myelinated, and all C-fibers are unmyelinated; A-fibers have faster conduction speeds and larger diameter axons as compared to C-fibers, but of the A-fibers only A-delta-fibers play a role in nociception.[1][3][4][7] The larger and faster mechanothermal A-delta-fibers are activated first when encountering a noxious stimulus, they provide what is known as "first" or fast pain.[1][3][4][7]
Calcium ion studies have shown that the severity of the noxious stimulus, whether it is thermal energy or mechanical force, correlates to the intensity of activation of the polymodal C-fibers.[3][4][5][7] C-fibers become stimulated secondary to A-delta-fibers, and only if the noxious stimulus is persistent, once activated, the C-fibers quickly adapt to the stimulus such that if the stimulus prolongs, then their reactivity is lost.[3][4][7] The sensation carried by A-delta-fibers has been described as "pricking" pain while sensation carried by C-fibers has conversely been described as "dull" or "pressing" pain.[4][7]
A final note is that while many electrophysiological studies have presented findings suggesting the vast majority of nociceptive fibers act in a polymodal fashion at all times, calcium ion studies have found that in vivo nociceptors often report only a single nociceptive modality, i.e., an Adelta-fiber reporting noxious heat but not noxious force.[5][7] The inherent polymodal functionality of nociceptors and nociceptive nerve fibers may be an artifact of the trauma and resultant inflammation-induced when conducting the more invasive electrophysiological studies; the resultant inflammation has been thought to sensitize the otherwise monomodal nociceptive neurons and activate their polymodal activity.[5] Noxious stimuli can induce monomodal or silent nociceptors to become active and polymodal, and such activation may be a regular motif that gets mirrored throughout virtually all innervated tissues.[7]
Cellular Level
RT-PCR has provided the means to discover and classify nociceptive neurons based on the specific receptors they possess, which are sensitive to noxious stimuli.[1] Nociceptors in the skin and other peripheral organs form highly complex and interconnected networks with each other. Schwann cells are peripheral glial cells that envelop these networks everywhere except where the terminal axons project through the basement membrane of the epidermis and the nerve endings become ‘free' or ‘naked' (i.e., unmyelinated).[3] Unfortunately, the complex and sequestered nature of nociceptors in vivo has made them difficult to study, and much of what researchers know about the activity, gene expression, and important factors present in nociceptors has been discovered by studying nociceptive neurons grown in culture.[3] While nociceptors grown in culture have been found to possess many of the same molecules found in vivo, it is not currently possible to discern the absence of any in vivo molecules that only receive the necessary signals produced when the nociceptive neuron is fully imbedded in cutaneous tissue.[3]
Polymodal Nociceptors: Thermal, Mechanical, Chemical
Thermal nociceptive receptors are a subdivision of the transient receptor potential cation channel (TRP) family of receptors. Of the TRP channel family, which is composed of several ligand-specific subfamilies, the vanilloid variant (TRPV) is found in thermal nociceptors and is responsible for the transduction of thermal stimuli. TRPV monomers form tetrameric structures that contain a central pore through the cell membrane, and these tetramers may be homo-tetrameric or hetero-tetrameric regarding their composition of the four TRPV subunits. TRPV1 expresses on heat-sensitive C-fibers and TRPV2 is expressed on heat-sensitive A-delta-fibers; these receptors are the primary noxious thermoreceptors, they are stimulated at 40 to 43 degrees Celsius and 52 degrees Celsius respectively.[1][3][4] It is important to note that these temperature-regulated receptors get dynamically set; while such high base-thresholds indicate inactivity under physiologic conditions, the temperatures necessary to stimulate TRPV channels may be significantly lowered during states of inflammation.[4][5][7][9] The two remaining members of the family, TRPV3 and TRPV4, function at 33 to 39 degrees Celsius and 27 to 34 degrees Celsius, respectively. Taken together, the TRPV family is capable of detecting a wide ride range of thermal energy levels that represents a range spanning normal, physiologic conditions through to those that are noxious and potentially harmful.
When TRPV1 and TRPV2 detect sufficiently high temperatures, they open and allow an influx of calcium, which depolarizes the neuron. Although TRPV1 and TRPV2 channels prefer calcium, by definition, they are non-selective cation channels.[1] Calcium-induced depolarization stimulates nearby voltage-gated sodium channels that generate an afferent action potential. In addition to detecting noxious thermal energy, TRPV receptors are also capable of detecting many chemical molecules, as well. TRPV chemical ligands include prostaglandins (proinflammatory), bradykinin (proinflammatory), capsaicin (natural product), anandamide (neurotransmitter), olvanil (antiinflammatory), resiniferatoxin (natural product), and acidic pH (protons).[1][3][7] TRPV sensitivity to capsaicin and acidic pH illustrates the polymodal sensitivity of nociceptors seen in vivo and may explain why such stimuli share the sensation of burning.[1][3] TRPV1 and TRPV2 are found throughout the central and peripheral nervous systems, as well as within the spleen and lung.[1] TRPV3 and TRPV4 are expressed as TRPV1 and TRPV2 but are also present in the skin, sperm, and many visceral organs.[11][12]
In comparison to the amount of information known about thermal nociceptors, very little is known regarding mechanical nociceptors and the transduction of noxious mechanical force. Provided mechanical nociceptors, also called mechanoreceptors, function similarly to other force-sensitive ion channels, it can be presumed that these channels are present and expressed on the free nerve endings that have penetrated the basement membrane of the epidermis – the unmyelinated nerve endings.[3] As explained previously regarding thermoreceptors, the sequestered nature of mechanoreceptors in vivo has prevented their careful study. Much of what is known has been gathered by studying nociceptive neurons grown in culture. While such studies have identified several mechano-sensitive ion channels, the currents produced by these channels are both nociceptive as well as anti-nociceptive.[3] Inhibitory mechanoreceptors, named TREK and Kv1.1, may play a role in keeping the threshold for noxious mechanical force sufficiently high to prevent overactivity of other mechanoreceptors, which is an idea somewhat analogous to TRPV3 and TRPV4 being responsible for lower thermal energy levels than TRPV1 and TRPV2.[1][3][9] One final hypothesis, yet to be studied in mammalian tissue, is that noxious mechanical forces break the delicate nociceptive nerve endings, and the release of intracellular contents such as ATP and neuropeptides lead to inflammation, neural depolarization, and sensation of pain.[3]
Chemical nociception refers to a nociceptive neuron that expresses receptors capable of detecting noxious, irritating, or harmful chemicals.[3] Since heat-sensitive TRPV receptor proteins are also capable of detecting many noxious chemicals, it should be no surprise that the TRP receptor family also function as chemical receptors. In terms of chemical sensitivity TRPV1 is surpassed by TRPA1, while ‘V' indicated vanilloid, ‘A' simply indicates subfamily A. TRPV1 is capable of binding capsaicin, prostaglandins, bradykinin, and other important pro-inflammatory molecules, however TRPA1 is capable of detecting a wide variety of oily and pungent isothiocyanate compounds such as mustard oils, cinnamaldehyde, and formaldehyde, which is commonly injected to study the activity of TRPA1 receptors, and is even responsible for mediating the stinging, tearful sensations induced by chopping onions or exposure to synthetic tear gases.[1][2][3] TRPA1 receptor activation also differs significantly from TRPV1 activation, whereas TRPV1 undergoes a conformational change when bound by its ligand, noxious chemicals bind to TRPA1 via a nucleophilic attack that produces covalent modification of cysteine residues on the intracellular portion of the receptor.[3] Once the intracellular modification has occurred, the receptor opens and depolarizes the neuron.[3] The acid-sensing ion channels (ASIC) are another example of a noxious chemical receptor, as their name implies ASICs are stimulated by protons that activate the receptor, inducing their cation channel to open.[5] ASIC receptors are found throughout the CNS and peripheral nervous system (PNS) and are capable of detecting acidic conditions of many different etiologies; these conditions span from the more benign lactic acidosis triggering muscle pain to the more severe acidosis produced by the tissue damage induced by ischemia, stroke, or tumor growth.[13]
Development
Nociceptive neurons arise from neural crest stem cells that have migrated out of the neural tube before the tube closed. More specifically, nociceptors develop from the dorsal population of neural crest stem cells within the neural crest tissue.[11] Although transcription factors necessary for nociceptive differentiation remain unknown, all nociceptive neurons express the TrkA receptor to nerve-growth factor.[11]
Organ Systems Involved
Nociceptive neurons are a component of the peripheral nervous system (PNS). In this section, we will evaluate how a noxious stimulus detected at the periphery is transmitted to the central nervous system (CNS). It is important to note that somatic and visceral nociceptive stimuli reach the higher-ordered brain centers via different routes. As will be covered more thoroughly, the skin and organs of locomotion receive their somatic sensory fibers from the spinal and trigeminal nerves alone. In contrast to the singular route of somatic structure innervation, the majority of the viscera connect to the CNS via two distinct avenues. By using different neural pathways, the sympathetic and parasympathetic divisions of the autonomic nervous system create the two routes for a visceral nociceptive signal to reach specific destinations in the brain.[1][7] Of the viscera, parasympathetic nociceptive fibers utilize specific cranial nerves, while sympathetic nociceptive fibers utilize either the communicating branches of the thoracic and lumbar nerves or a select set of cranial nerves.[1]
A further distinction comes from the modulation of nociceptive signals as they ascend towards the brain. While local signals may modulate somatic nociception all along the polyneuronal chain, it is the substantia gelatinosa of the spinal cord dorsal horn, the spinal nucleus of the trigeminal nerve, or several descending pathways that primarily act to facilitate or inhibit the somatic nociceptive signals.[1][4][6]
This section will conclude with a brief but closer look at the previously mentioned polyneuronal trails that connect peripheral nociceptive nerve endings to the spinal cord, and then onwards to the brain. All primary afferent nociceptive nerve fibers project from their peripheral locations towards the spinal cord. If the primary afferent fiber is large and high-threshold, such as A-delta-fibers, it will project into the superficial dorsal horn of the spinal cord. This area is also referred to as Rexed laminae I and II.[7] However, if the primary afferent is large but low-threshold, such as A-beta-fibers, it will instead project through the dorsal roots, eventually terminating in the deeper lamina (Lamina III-V).[7] Both neurons synapse in their respective lamina, from there second-order dorsal horn neurons, carry the signal onward.[7] Depending on the synapsing neuron's type and location, its signal will get carried by one of two possible types of dorsal horn neurons, those that are "nociceptive specific" and those that are of wide dynamic range.[7]
Secondary "nociceptive specific" neurons arise from the nociceptive Adelta-fibers that synapsed in the superficial dorsal horn of the spinal cord, within laminae I and II.[1][7] The other population of secondary neurons, those of wide dynamic range, synapse with A-beta-, A-delta-, and C-fibers deeper in the dorsal horn of the spinal cord, laminae III-V.[1][7] The convergence of signals from primary A-beta-, A-delta-, and C-fibers in the deeper laminae defines why these secondary neurons are referred to as wide dynamic range neurons; they are capable of transmitting the graded potentials produced by both sub-nociceptive and nociceptive stimuli.[7] In both populations of secondary dorsal horn neurons, the excitatory neurotransmitter glutamate gets released from the primary fibers, which are detected by glutamatergic receptors (i.e., AMPA) on the dendrites of the secondary fibers.
The secondary projection fibers go on to ascend the spinal cord and cross through the ventrolateral tracts before finally reaching their supraspinal targets.[7] Generally, all projection neurons that reach the brain by way of the ventrolateral tracts diverge into two distinct trajectories.[7] These distinct trajectories differentiate whether the carried stimulus will ultimately provide the location and intensity of the stimulus or become integrated into an emotional response and change in affect.[1][7] Following the first path, for a stimulus to provide location and intensity information, it must exit the ventrolateral tracts of the spinal cord and then enter the somatosensory thalamus. From there, a tertiary neuron will carry the signal to its final destination in the somatosensory cortex.[1][7] Regarding the second path, if a stimulus requires integration into emotion, it must exit the ventrolateral tracts of the spinal cord and synapse with tertiary neurons in the medial and ventromedial thalamus.[7] The tertiary neurons exit the thalamus and terminate in the inferior insula or the anterior cingulate nucleus, both of which are in the limbic forebrain.[1][7]
Mechanism
The general mechanism by which a nociceptive neuron detects and reacts to a noxious stimulus has been a focus throughout this article. However, herein is a closer look at a model nociceptor, in this case, a thermosensitive nociceptor, with the goal being to better explain the cell-specific activities regarding detection, activation, afferent signaling, local signaling, and signal termination.
A noxious heat stimulus (if under physiologic conditions) or a sub-noxious heat stimulus (if sensitized by inflammatory markers such as prostaglandins or bradykinin) gets detected by TRPV1 (40 to 43 degrees Celsius) or TRPV2 (52 degrees Celsius) receptors.[1][3][7] The heat produces a conformational change that opens the non-selective cation (Ca2+) channel, which produces a local depolarization. The depolarization gets propagated by the opening of voltage-gated sodium channels, which generates an afferent action potential that travels to the brain.
The initial depolarization also mediates the release of many pro-inflammatory cytokines into the extracellular milieu; these cytokines include substance P and neurokinin A, which are neuropeptides, as well as the calcitonin-gene-related-peptide (CGRP).[1][3] The initial release of pro-inflammatory cytokines leads to a state of neuroinflammation. Neuroinflammation refers to a complex inflammatory process that originates with the activation of nociceptive neurons but ultimately involves every sensitive cell in the local environment.[3] The pro-inflammatory cytokines activate the surrounding immune cells, smooth muscle cells, epithelium, and endothelium, inducing them to release additional pro-inflammatory cytokines; IL-1b, IL-6, IL-8, TNFalpha, and extracellular ATP.[1][3][7][14] The responding pro-inflammatory signals further reinforce and spread the neuroinflammation.[1][3][7][14] On the surface of the skin, neuroinflammation produces visible redness and swelling or ‘flare' due to the vasodilation and increased permeability of local blood vessels, which allow for the extravasation of plasma into the extracellular tissues where it pools and collects.[3]
The activated channels close once they become dephosphorylated, or the noxious stimulus ceases. Alternatively, only the overwhelming influx of calcium and sodium cations may halt the local inflammatory and nociceptive signals.[1][15] Calcium overload in nociceptive neurons depletes the original neuropeptides, inhibits anterograde axonal trafficking of new neuropeptides, and causes the cutaneous nerve ending to reversibly collapse and retract from the periphery.[1][15] The TRPV cation channel then enters a lengthy refractory period while the intracellular calcium gradient gets restored, and the conformation changes in the receptor protein, and the peripheral portion of the axon itself reverses.[1][15]
Pathophysiology
Nociceptive neurons are dedicated to detecting noxious stimuli and alerting the brain when an injury is likely or imminent. Since pain is such an essential homeostatic mechanism for preserving health and avoiding morbidity, the person cannot easily ignore these sensations initially detected by nociceptive neurons or neural circuits. When nociceptors, or their secondary neural circuitry, are overstimulated, they are capable of producing disabling sensations of pain and damage in otherwise healthy tissues. Often, the increased activity of otherwise-unaffected nociceptors is physiologic; such is the case when an inflammatory state may sensitize thermal nociceptors and allow them to depolarize upon contact with sub-noxious temperatures.
Allodynia refers to a state where nociceptors have become indiscriminately sensitized, such that they depolarize in the presence of sub-noxious stimuli.[3][4] Regardless of the aberrant source of the nociceptive signals reaching the brain, the brain interprets the signals as allodynia, the unprovoked sensation of pain and damage. Allodynia is distinct from hyperalgesia. Hyperalgesia refers to a painful stimulus producing an exaggerated pain response well above the level of pain normally expected from such a stimulus.[3][4] Alternatively, "wind-up" is a measurable phenomenon present in nociceptive C-fibers, but not nociceptive A-fibers, where a small, moderately fast, a constant and repetitive string of stimuli produce larger and larger depolarizations of the activated nerve fiber.[7] The moderately high-frequency stimulus increases the magnitude of depolarization by maintaining the nerve fiber in a state of partial depolarization; the membrane potential is much closer to the threshold for depolarization than the normal resting potential.[7] Thus, each subsequent stimulus depolarizes the membrane to a greater extent than the last. The greater the depolarization, the more local voltage-gated sodium channels get recruited, and the intensity of the afferent action potential generated becomes greater.[7] Separate from both allodynia and hyperalgesia, injury-induced neuromodulatory changes in the spinal cord can allow afferent signals from neurons sensitive to far more innocuous stimuli, i.e., light touch or lower temperatures, to gain access to the same spinal tracts nociceptive signals use to reach the brain.[3] When these innocuous stimuli reach the brain, they get interpreted as if they originated from nociceptors. Such aberrant signals can produce debilitating intense noxious pain.[3]
Clinical Significance
Every day, patients seek medical care following injuries involving high heat, extreme cold, sizeable mechanical force, or noxious chemical exposure. Patients are only aware of their injuries because of the functional nociceptors found throughout their bodies. Nociceptors transition acute pain into inflammatory pain when the duration of stimulus persists, and nociceptors release their pro-inflammatory markers, sensitizing local, responsive cells.[7][8] Iatrogenic causes of nociceptive pain are not difficult to predict. Thermal nociceptive activation may be induced purposefully by electrical ablation, or accidentally by metal surgical instruments being utilized too soon after steam sterilization. Physical nociceptive activation may occur in response to damage via scalpels, curettes, drills, and more. Chemical nociceptive activation may get induced following the life-threatening surge in cellular waste products released into a patient's bloodstream during tumor lysis syndrome, or by the more benign muscle cramps that occur during anaerobic exercise producing a local lactic acidosis. The previously mentioned sources stimulate nociceptive pain, while neuropathic pain arises from diseases or damage mediated directly to sensory nerves. Sources of neuropathic pain include diabetic neuropathy, shingles, or postherpetic neuralgia.[2][6][8] Inactive nociceptors may guide conscious actions and motivate pain and injury avoidance.[4][9] The findings of three poignant studies demonstrate this theory; patients with painless channelopathies, patients rendered insensitive to pain due to leprosy, and patients with painless Charcot joints of tabes dorsalis.[9] The results of a lack of nociception are difficult to imagine, but such scenarios merit consideration in patients with an explicable lack of awareness following serious injuries. Finally, it is worth reiterating that allodynia refers specifically to nociceptors that have become indiscriminately sensitized and the discomfort or disability that is produced by this sensitization.[3][4]
Media
(Click Image to Enlarge)
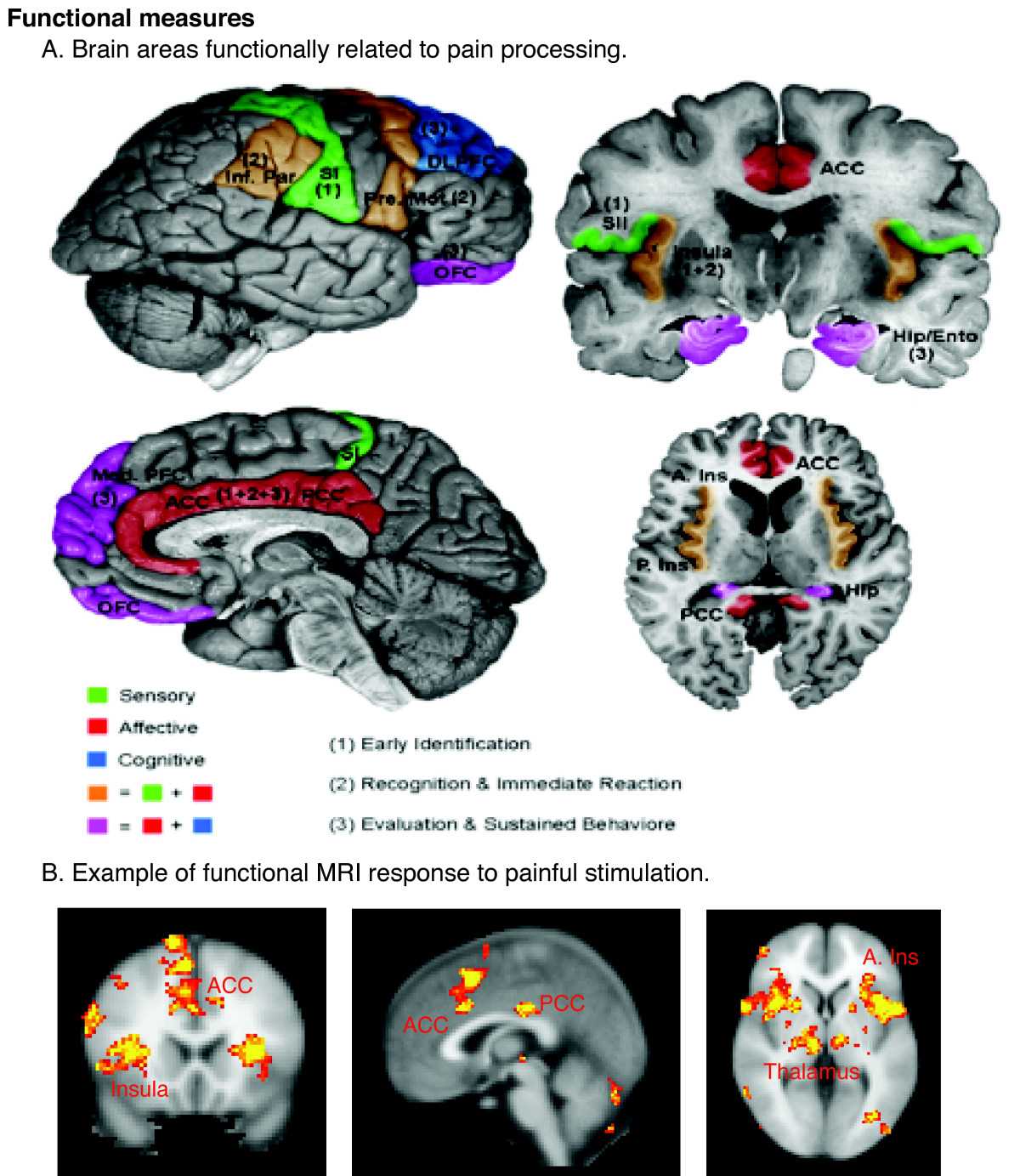
Regions of the cerebral cortex associated with pain. Neuroimaging revolutionizes therapeutic approaches to chronic pain Contributed by Wikimedia Commons, Borsook D, Moulton EA, Schmidt KF, Becerra LR (CC BY 2.0)
(Click Image to Enlarge)
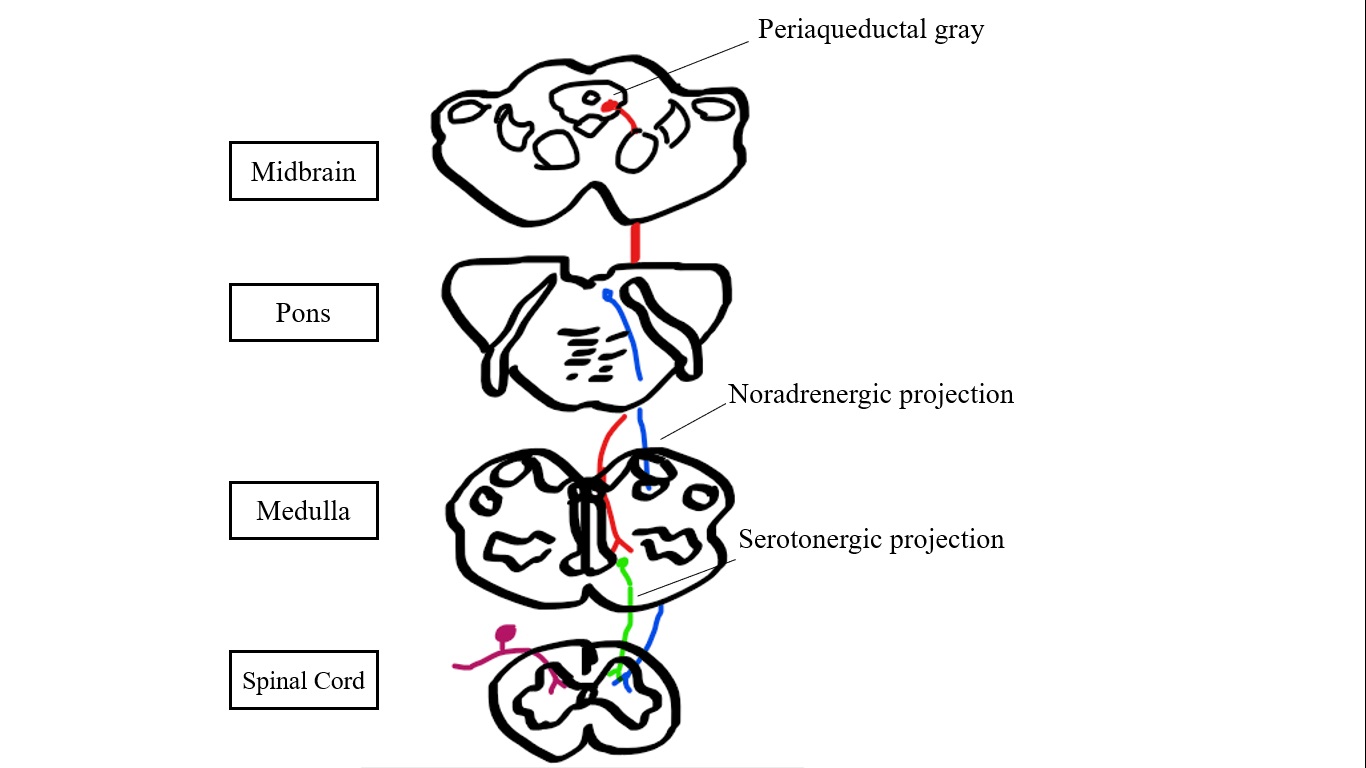
Periaqueductal pain pathways, sketched Sketched by Miriam Mokhtar MD
References
Frias B, Merighi A. Capsaicin, Nociception and Pain. Molecules (Basel, Switzerland). 2016 Jun 18:21(6):. doi: 10.3390/molecules21060797. Epub 2016 Jun 18 [PubMed PMID: 27322240]
Ang ST, Ariffin MZ, Khanna S. The forebrain medial septal region and nociception. Neurobiology of learning and memory. 2017 Feb:138():238-251. doi: 10.1016/j.nlm.2016.07.017. Epub 2016 Jul 19 [PubMed PMID: 27444843]
Tracey WD Jr. Nociception. Current biology : CB. 2017 Feb 20:27(4):R129-R133. doi: 10.1016/j.cub.2017.01.037. Epub [PubMed PMID: 28222285]
Sneddon LU. Comparative Physiology of Nociception and Pain. Physiology (Bethesda, Md.). 2018 Jan 1:33(1):63-73. doi: 10.1152/physiol.00022.2017. Epub [PubMed PMID: 29212893]
Level 2 (mid-level) evidenceSt John Smith E. Advances in understanding nociception and neuropathic pain. Journal of neurology. 2018 Feb:265(2):231-238. doi: 10.1007/s00415-017-8641-6. Epub 2017 Oct 14 [PubMed PMID: 29032407]
Level 3 (low-level) evidenceMitsi V, Zachariou V. Modulation of pain, nociception, and analgesia by the brain reward center. Neuroscience. 2016 Dec 3:338():81-92. doi: 10.1016/j.neuroscience.2016.05.017. Epub 2016 May 14 [PubMed PMID: 27189881]
Woller SA, Eddinger KA, Corr M, Yaksh TL. An overview of pathways encoding nociception. Clinical and experimental rheumatology. 2017 Sep-Oct:35 Suppl 107(5):40-46 [PubMed PMID: 28967373]
Level 3 (low-level) evidenceFalk S, Dickenson AH. Pain and nociception: mechanisms of cancer-induced bone pain. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2014 Jun 1:32(16):1647-54. doi: 10.1200/JCO.2013.51.7219. Epub 2014 May 5 [PubMed PMID: 24799469]
Level 3 (low-level) evidenceBaliki MN, Apkarian AV. Nociception, Pain, Negative Moods, and Behavior Selection. Neuron. 2015 Aug 5:87(3):474-91. doi: 10.1016/j.neuron.2015.06.005. Epub [PubMed PMID: 26247858]
Bista P, Imlach WL. Pathological Mechanisms and Therapeutic Targets for Trigeminal Neuropathic Pain. Medicines (Basel, Switzerland). 2019 Aug 22:6(3):. doi: 10.3390/medicines6030091. Epub 2019 Aug 22 [PubMed PMID: 31443547]
Cheng W, Yang F, Takanishi CL, Zheng J. Thermosensitive TRPV channel subunits coassemble into heteromeric channels with intermediate conductance and gating properties. The Journal of general physiology. 2007 Mar:129(3):191-207 [PubMed PMID: 17325193]
Vennekens R, Owsianik G, Nilius B. Vanilloid transient receptor potential cation channels: an overview. Current pharmaceutical design. 2008:14(1):18-31 [PubMed PMID: 18220815]
Level 3 (low-level) evidenceCheng YR, Jiang BY, Chen CC. Acid-sensing ion channels: dual function proteins for chemo-sensing and mechano-sensing. Journal of biomedical science. 2018 May 24:25(1):46. doi: 10.1186/s12929-018-0448-y. Epub 2018 May 24 [PubMed PMID: 29793480]
Miller RE, Miller RJ, Malfait AM. Osteoarthritis joint pain: the cytokine connection. Cytokine. 2014 Dec:70(2):185-93. doi: 10.1016/j.cyto.2014.06.019. Epub 2014 Jul 24 [PubMed PMID: 25066335]
Level 3 (low-level) evidenceAnand P, Bley K. Topical capsaicin for pain management: therapeutic potential and mechanisms of action of the new high-concentration capsaicin 8% patch. British journal of anaesthesia. 2011 Oct:107(4):490-502. doi: 10.1093/bja/aer260. Epub 2011 Aug 17 [PubMed PMID: 21852280]
Level 3 (low-level) evidence