Introduction
Immunoglobulin A (IgA) pemphigus, a rare autoimmune blistering disease, manifests as painful and pruritic vesiculopustular eruptions. These eruptions occur due to circulating IgA antibodies targeting keratinocyte cell surface components involved in cell-to-cell adherence. Although associated with various malignancies and chronic conditions, its precise etiology remains unclear. IgA pemphigus comprises 2 distinct subtypes: subcorneal pustular dermatosis and intraepidermal neutrophilic dermatosis. The subcorneal pustular dermatosis subtype presents with intercellular IgA deposition against desmocollin-1 glycoprotein, primarily in the upper epidermis, whereas the intraepidermal neutrophilic dermatosis subtype exhibits autoantibodies targeting desmoglein members in the cadherin superfamily, predominantly in the lower epidermis.[1][2] Although IgA pemphigus typically has a milder disease course compared to IgG-driven pemphigus, it necessitates careful diagnosis and aggressive treatment with steroids and dapsone to prevent recurrence.
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
Antikeratinocyte cell surface autoantibodies are responsible for the disease process associated with IgA pemphigus, but the inciting mechanism is unknown. Research suggests the potential involvement of interleukin 5 (IL-5), a type 2 T-helper cytokine associated with stimulating IgA class antibody and γδ T-cell receptor–containing T cells, crucial for mucosal IgA production. Moreover, the IgA autoantibodies possess specific binding sites for the monocyte/granulocyte IgA-Fc receptor (CD89), which may allow neutrophils to accumulate intraepidermally, causing the blistering process to occur.
IgA pemphigus is associated with monoclonal IgA gammopathy and multiple myeloma.[3] Experts are uncertain if monoclonal gammopathy precedes or follows IgA pemphigus, but most cases are present at the time of diagnosis. Other associated diseases include HIV infection, Sjögren syndrome, rheumatoid arthritis, lung cancer, ulcerative colitis, peripheral T-cell lymphoma, chronic myeloid leukemia, and diffuse large B-cell lymphoma. Although the direct relationship between the various diseases and IgA pemphigus is still unclear, healthcare professionals should thoroughly diagnose patients presenting with IgA pemphigus for hematologic, gastrointestinal, rheumatological, and infectious disorders. [4][5]
Epidemiology
IgA pemphigus stands out as one of the most uncommon types of autoimmune blistering diseases, with a limited understanding of its epidemiology, including age and race demographics. Although it can manifest at any age, the reported range is 1 month to 94 years, predominantly emerging in adulthood, with the average onset occurring between the fourth and sixth decades.[6] Notably, no distinct gender predilection is apparent in reported cases.[7]
Pathophysiology
The pathogenesis of IgA pemphigus involves IgA autoantibodies that specifically target desmosomal and nondesmosomal keratinocyte cell surface components, notably desmoglein-1, desmoglein-3, and desmocollin-1. These molecules are integral to cell-to-cell adherence and belong to the cadherin superfamily of glycoproteins. In the subcorneal pustular dermatosis subtype, desmocollin-1 serves as the antigen. In contrast, in the intraepidermal neutrophilic dermatosis subtype, interactions occur with both desmoglein-1 and desmoglein-3.[8][9] The binding of IgA antibodies to the IgA-Fc receptor triggers an intense inflammatory response, leading to epidermal neutrophilic infiltration followed by the formation of blisters and pustules. Despite identifying IgA antibody targets, the precise mechanistic cascade underlying the clinical presentation of IgA pemphigus remains elusive and necessitates further research.[1]
Histopathology
Histological examination of IgA pemphigus reveals subcorneal blisters with massive neutrophilic infiltration and a mild loss of cohesion between keratinocytes. This histopathological analysis aids in distinguishing the 2 major subtypes of IgA pemphigus. The subcorneal pustular dermatosis subtype may reveal minimal subcorneal acantholysis and pustules with increased IgA autoantibody intensity on the epidermis's upper surface. In contrast, the characteristic features of the intraepidermal neutrophilic dermatosis subtype are pustules deeper in the epidermis and inflammatory infiltrates, primarily in the entire or lower part of the dermis (see Image. Intraepidermal Neutrophilic IgA Pemphigus).[10][11] Although IgA is predominately detected on direct immunofluorescence, occasionally, IgG and complement component C3 may be detected to a much lesser extent. Unlike IgG pemphigus, acantholysis may be minimal or absent in IgA pemphigus.
History and Physical
When obtaining a patient’s medical history, clinicians should ascertain the presence of mucosal involvement, as the presence of mucosal lesions can help distinguish between different subtypes of pemphigus disease. IgA pemphigus lacks mucosal involvement. The initial manifestation of IgA pemphigus involves a subacute onset of flaccid blisters on an erythematous base filled with clear fluid. These evolve into pustules that rapidly rupture, forming annular crusts. Patients often describe these plaques as both painful and pruritic. Although commonly observed in flexural areas such as the axilla and groin, the trunk and extremities are the most commonly affected areas.[12] Patients typically experience symptoms limited to skin manifestations, with no systemic symptoms such as fever, malaise, headache, or weight loss.[13][14]
Evaluation
The evaluation of suspected IgA pemphigus begins with a skin biopsy. Clinicians use histopathology and direct and indirect immunofluorescence techniques to establish the diagnosis. Clinicians should obtain a 4-mm lesional biopsy from the edge of an early lesion or erosion for hematoxylin and eosin staining and routine histopathological examination. For direct immunofluorescence, an additional perilesional skin biopsy should be taken from unaffected skin, situated 4 mm away from a vesicle or erosion. Lesional skin biopsies for direct immunofluorescence are more likely to give a false negative result due to the inflammatory response potentially destroying the immunoreactants.
Histological examination reveals the hallmark of intraepidermal neutrophilic infiltration and the additional classic histopathological changes (refer to the Histology section for more information on the histopathology of IgA pemphigus).[15][16] As acantholysis is minimal in IgA pemphigus, direct immunofluorescence may be considered an early screening tool for diagnosing IgA pemphigus in patients with diffuse pustular eruptions. Direct immunofluorescence allows clinicians to detect the absence or presence of IgA autoantibodies on epidermal cell surfaces. Indirect immunofluorescence detects the presence of IgA circulating autoantibodies in patient serum using patient serum on monkey esophagus or other epithelial substrates. Indirect immunofluorescence detects circulating IgA autoantibodies in approximately 50% of affected patients.[1]
Immunoblotting allows clinicians to document the specific skin antigen recognized by the patient's IgA autoantibodies. The sensitivity of immunoblotting is approximately 40% in revealing IgA reactivity to any autoantigen. Enzyme-linked immunosorbent assay can detect specific desmosomal antigens in patients with IgA pemphigus but is associated with a low sensitivity of approximately 55%.
Treatment / Management
Due to the inflammatory nature of the disease, the primary treatment approach for IgA pemphigus involves the use of oral and topical corticosteroids with a suggested daily dose of 0.5 to 1 mg/kg.[17] However, patients should be aware of the potential adverse effects associated with the long-term use of steroids, including osteoporosis, diabetes, cataracts, adrenal suppression, and infection. In contrast to IgG pemphigus, IgA pemphigus typically does not respond adequately to steroid therapy alone.[18] Numerous studies demonstrate that combining dapsone with corticosteroids yields significantly better outcomes. Dapsone primarily works by suppressing neutrophilic infiltration. Healthcare professionals should carefully monitor patients receiving dapsone for potential adverse effects, such as hemolysis and methemoglobinemia.(B3)
Other medications reported to effectively treat IgA pemphigus include colchicine, retinoids, mycophenolate mofetil, and adalimumab.[19][20] Many of these medications were tested based on their successful treatment of other forms of neutrophilic dermatoses and classic pemphigus. For example, adalimumab, a recombinant human immunoglobulin antibody, is believed to be therapeutic due to its tumor necrosis factor-α (TNF-α) inhibition. TNF-α activates neutrophilic infiltration in the epidermis; thus, its inhibition may hinder the further progression of IgA pemphigus.[21] Rituximab, a monoclonal antibody targeting the B-lymphocyte antigen CD20 (CD-20) protein on cells, has also been safely used to treat patients with IgA pemphigus.[22](A1)
Clinicians should also consider providing proton-pump inhibitors and bisphosphonate therapy to prevent peptic ulceration and osteoporosis. Patients should receive counseling regarding weight-bearing exercise and adequate calcium and vitamin D intake. Patients should undergo a thorough physical examination and, when clinically indicated, appropriate testing for concurrent malignancies, inflammatory bowel disease, rheumatological diseases, and infectious diseases.
Differential Diagnosis
Due to the rarity of IgA pemphigus, clinicians should consider various differential diagnoses when managing and treating the condition. The clinical presentation of these conditions is remarkably similar, and careful investigation using histology and immunofluorescence may be necessary to differentiate the conditions.[13] The following are potential differential diagnoses for IgA pemphigus.
- Bullous impetigo
- Pustular psoriasis
- Linear IgA bullous dermatosis
- Dermatitis herpetiformis
- Subcorneal pustular dermatosis or Sneddon-Wilkinson disease
- Pemphigus foliaceus
Classic subcorneal pustular dermatosis, also known as Sneddon-Wilkinson disease, is a chronic dermatosis characterized by sterile pustular lesions that erupt in cyclical patterns. Similar to the subcorneal pustular dermatosis subtype of IgA pemphigus, the pustular lesions observed in Sneddon-Wilkinson disease coalesce in an annular pattern and eventually burst to form crusted plaques. The 2 conditions have the same distribution area, favoring the groin, trunk, and axillae while avoiding mucosal surfaces. Histological examination of Sneddon-Wilkinson disease demonstrates perivascular infiltration of neutrophils and mild spongiosis. However, in contrast to IgA pemphigus, direct immunofluorescence of Sneddon-Wilkinson disease reveals a negative result for IgA deposits against adhesion molecules such as desmocollin-1.[23]
Pemphigus foliaceus is characterized by flaccid bullae typically found on the trunk that eventually crust over, much like the lesions in IgA pemphigus. Generally considered a benign disease, pemphigus foliaceus responds well to topical and oral corticosteroids. A clinical differentiation between IgA pemphigus and pemphigus foliaceus is nearly impossible. Thus, immunofluorescence is critical in diagnosis. Direct immunofluorescence of pemphigus foliaceus demonstrates IgG autoantibodies against desmoglein-1 in contrast to the IgA deposits against desmocollin-1 found in IgA pemphigus. Therefore, proper histology and immunofluorescence diagnosis are essential in differentiating the 2 conditions.[24]
Prognosis
In contrast to classic pemphigus, IgA pemphigus typically manifests as a less severe and more localized condition. With appropriate treatment and regular monitoring, IgA pemphigus commonly resolves without scarring. Studies indicate that abruptly discontinuing oral steroids may lead to lesion recurrence; therefore, clinicians should gradually reduce and taper the dosage. Prognosis in cases of IgA pemphigus associated with other conditions such as malignancies, gastrointestinal diseases, or monoclonal gammopathy depends on the progression of the underlying conditions.[1][25]
Complications
Many potential complications associated with IgA pemphigus are due to the effects of long-term treatment. Secondary infection of the lesions and scarring are the primary complications associated with IgA pemphigus. The following are potential complications related to corticosteroid use.
- Osteoporosis
- Peptic ulcer disease
- Adrenal insufficiency
- Infection
- Growth restriction in children
- Weight gain
- Anemia
- Hypertension
- Diabetes [1]
Potential complications related to dapsone use include:
- Methemoglobinemia
- Hemolytic anemia
- Neutropenia
- Agranulocytosis
- Hepatic failure
- Drug rash with eosinophilia and systemic symptoms (DRESS) syndrome
Deterrence and Patient Education
IgA pemphigus is a rare autoimmune blistering disease that can cause painful and pruritic skin eruptions. These eruptions result from the body's immune system mistakenly attacking certain skin proteins. Unlike some other forms of pemphigus, IgA pemphigus usually presents as a milder condition with limited skin involvement.[26] Affected patients are at risk of secondary infection of open wounds and scarring. Pruritus is a prominent symptom; patients may scratch the lesions, increasing the risk of secondary infection. Patients should be aware that they must seek medical attention if they notice increased erythema, fevers, or purulent drainage.
Although IgA pemphigus typically heals without scarring, patients must understand the medications used for treatment, including any potential adverse effects. Clinicians should stress the importance of continuing treatment and regularly attending scheduled follow-up appointments. Abruptly stopping medications, especially oral steroids, can lead to a recurrence of skin lesions and adrenal insufficiency. Patients must understand the need for a gradual reduction in medication dosage. In addition, if concurrent medical conditions are present, such as malignancies or gastrointestinal diseases, effectively diagnosing and managing these conditions are essential for overall health and may impact the prognosis of IgA pemphigus.
Pearls and Other Issues
A newer classification of IgA pemphigus exists, describing 5 subtypes as follows:
- Subcorneal pustular dermatosis type IgA pemphigus
- Intraepidermal neutrophilic type IgA pemphigus
- IgA-pemphigus vegetans
- IgA-pemphigus vulgaris
- Unclassified IgA pemphigus
Pemphigus vegetans is a rare variant of pemphigus vulgaris. Clinicians classify patients with vegetating lesions and histology similar to pemphigus vegetans but with IgA antibodies as IgA pemphigus vegetans (IgA-PVeg). Those with desmoglein-1 or desmoglein-3 target antigens are diagnosed with having IgA pemphigus foliaceus (IgA-PF) or IgA pemphigus vulgaris (IgA-PV), respectively. The remainder that do not meet these criteria are diagnosed as unclassified or atypical IgA dermatosis.
Enhancing Healthcare Team Outcomes
IgA pemphigus is a rare autoimmune blistering disease characterized by painful and pruritic vesiculopustular eruptions resulting from IgA antibodies targeting keratinocyte cell surface components involved in cell-to-cell adherence. Two distinct forms of IgA pemphigus exist, both clinically similar but with distinct autoantibody target proteins. Although associated with various malignancies and chronic conditions, its exact cause remains unclear. Although typically milder than IgG-driven pemphigus, IgA pemphigus requires aggressive treatment with steroids and dapsone to prevent a recurrence. Collaborative healthcare teams should monitor patients for adverse effects and assess for concurrent conditions, ensuring tailored management.
Physicians, advanced care practitioners, nurses, pharmacists, and all other healthcare professionals caring for patients with IgA pemphigus must work collaboratively, each contributing their unique expertise. Seamless interprofessional communication is crucial for prompt information sharing and care coordination, optimizing patient outcomes and quality of life.[18] This coordination minimizes errors, reduces delays, and enhances patient safety, leading to improved team performance and patient-centered care that prioritizes improving immediate and long-term outcomes.
Media
(Click Image to Enlarge)
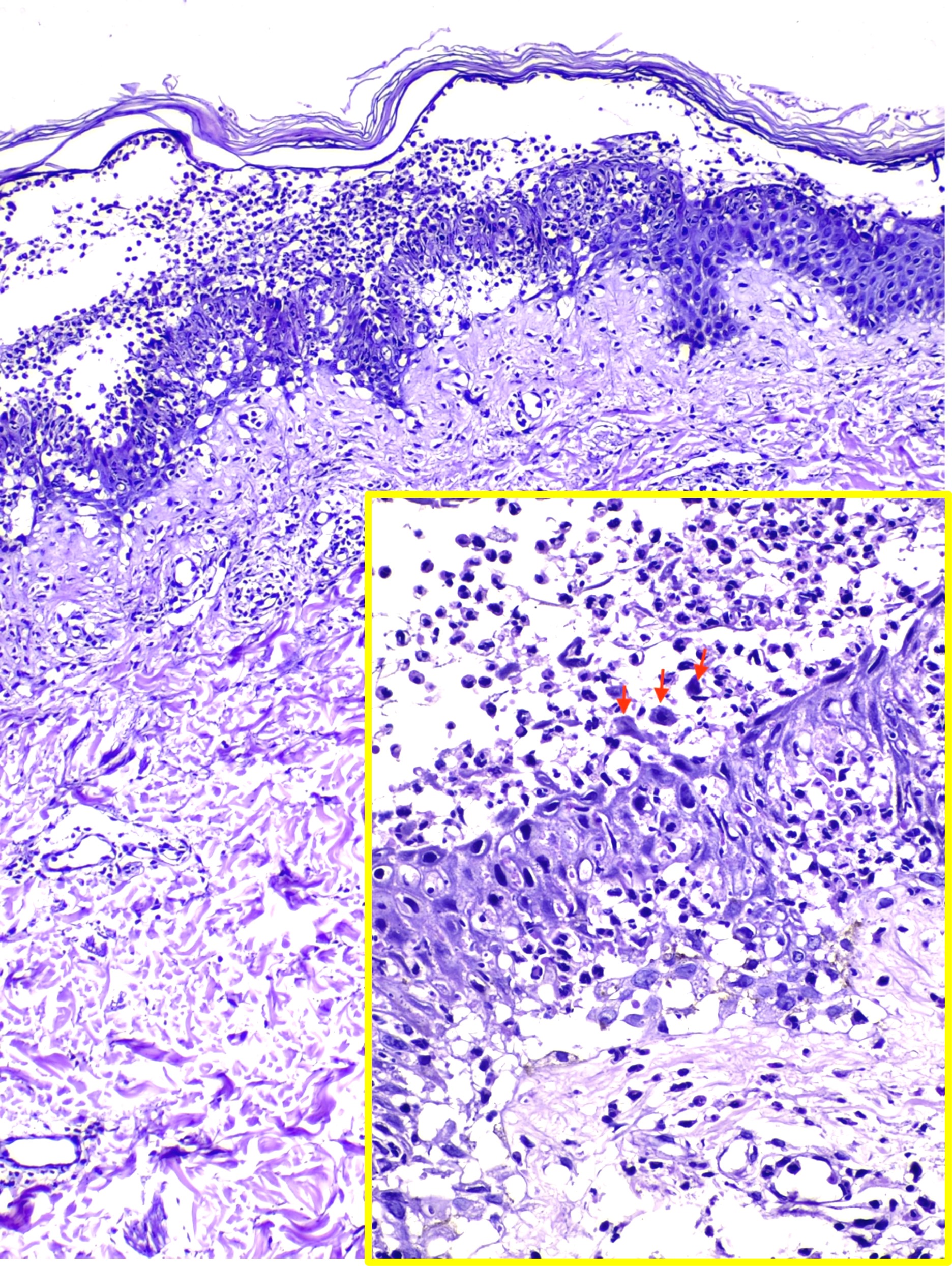
Intraepidermal Neutrophilic IgA Pemphigus. Hematoxylin and eosin staining of intraepidermal neutrophilic IgA pemphigus.
Contributed by M Abdel-Halim Ibrahim, MD
References
Porro AM, Caetano Lde V, Maehara Lde S, Enokihara MM. Non-classical forms of pemphigus: pemphigus herpetiformis, IgA pemphigus, paraneoplastic pemphigus and IgG/IgA pemphigus. Anais brasileiros de dermatologia. 2014 Jan-Feb:89(1):96-106. doi: 10.1590/abd1806-4841.20142459. Epub [PubMed PMID: 24626654]
Hegazy S, Bouchouicha S, Khaled A, Laadher L, Sellami MK, Zeglaoui F. IgA pemphigus showing IgA antibodies to desmoglein 1 and 3. Dermatology practical & conceptual. 2016 Oct:6(4):31-33 [PubMed PMID: 27867744]
Wallach D. Intraepidermal IgA pustulosis. Journal of the American Academy of Dermatology. 1992 Dec:27(6 Pt 1):993-1000 [PubMed PMID: 1479108]
Kridin K, Patel PM, Jones VA, Cordova A, Amber KT. IgA pemphigus: A systematic review. Journal of the American Academy of Dermatology. 2020 Jun:82(6):1386-1392. doi: 10.1016/j.jaad.2019.11.059. Epub 2019 Dec 5 [PubMed PMID: 31812619]
Level 1 (high-level) evidenceKridin K, Schmidt E. Epidemiology of Pemphigus. JID innovations : skin science from molecules to population health. 2021 Mar:1(1):100004. doi: 10.1016/j.xjidi.2021.100004. Epub 2021 Feb 20 [PubMed PMID: 34909708]
Cheng HF, Tsoi WK, Ng MMT, Ip WK, Ho KM. IgG/IgA pemphigus with differing regional presentations. JAAD case reports. 2022 Oct:28():119-122. doi: 10.1016/j.jdcr.2022.03.026. Epub 2022 Apr 1 [PubMed PMID: 36147207]
Level 3 (low-level) evidenceHashimoto T, Teye K, Hashimoto K, Wozniak K, Ueo D, Fujiwara S, Inafuku K, Kotobuki Y, Jukic IL, Marinović B, Bruckner A, Tsuruta D, Kawakami T, Ishii N. Clinical and Immunological Study of 30 Cases With Both IgG and IgA Anti-Keratinocyte Cell Surface Autoantibodies Toward the Definition of Intercellular IgG/IgA Dermatosis. Frontiers in immunology. 2018:9():994. doi: 10.3389/fimmu.2018.00994. Epub 2018 May 7 [PubMed PMID: 29867971]
Level 3 (low-level) evidenceYasuda H, Kobayashi H, Hashimoto T, Itoh K, Yamane M, Nakamura J. Subcorneal pustular dermatosis type of IgA pemphigus: demonstration of autoantibodies to desmocollin-1 and clinical review. The British journal of dermatology. 2000 Jul:143(1):144-8 [PubMed PMID: 10886149]
Level 3 (low-level) evidenceAmagai M. Adhesion molecules. I: Keratinocyte-keratinocyte interactions; cadherins and pemphigus. The Journal of investigative dermatology. 1995 Jan:104(1):146-52 [PubMed PMID: 7798634]
Robinson ND, Hashimoto T, Amagai M, Chan LS. The new pemphigus variants. Journal of the American Academy of Dermatology. 1999 May:40(5 Pt 1):649-71; quiz 672-3 [PubMed PMID: 10321591]
Amagai M. Autoimmunity against desmosomal cadherins in pemphigus. Journal of dermatological science. 1999 Jun:20(2):92-102 [PubMed PMID: 10379702]
Level 3 (low-level) evidenceMalik AM, Tupchong S, Huang S, Are A, Hsu S, Motaparthi K. An Updated Review of Pemphigus Diseases. Medicina (Kaunas, Lithuania). 2021 Oct 9:57(10):. doi: 10.3390/medicina57101080. Epub 2021 Oct 9 [PubMed PMID: 34684117]
Kridin K. Pemphigus group: overview, epidemiology, mortality, and comorbidities. Immunologic research. 2018 Apr:66(2):255-270. doi: 10.1007/s12026-018-8986-7. Epub [PubMed PMID: 29479654]
Level 3 (low-level) evidenceHashimoto T, Yasumoto S, Nagata Y, Okamoto T, Fujita S. Clinical, histopathological and immunological distinction in two cases of IgA pemphigus. Clinical and experimental dermatology. 2002 Nov:27(8):636-40 [PubMed PMID: 12472534]
Level 3 (low-level) evidenceTsuruta D, Ishii N, Hamada T, Ohyama B, Fukuda S, Koga H, Imamura K, Kobayashi H, Karashima T, Nakama T, Dainichi T, Hashimoto T. IgA pemphigus. Clinics in dermatology. 2011 Jul-Aug:29(4):437-42. doi: 10.1016/j.clindermatol.2011.01.014. Epub [PubMed PMID: 21679872]
Toosi S, Collins JW, Lohse CM, Wolz MM, Wieland CN, Camilleri MJ, Bruce AJ, McEvoy MT, Lehman JS. Clinicopathologic features of IgG/IgA pemphigus in comparison with classic (IgG) and IgA pemphigus. International journal of dermatology. 2016 Apr:55(4):e184-90. doi: 10.1111/ijd.13025. Epub 2015 Nov 13 [PubMed PMID: 26566588]
Camisa C, Warner M. Treatment of pemphigus. Dermatology nursing. 1998 Apr:10(2):115-8, 123-31 [PubMed PMID: 9697444]
Hirata Y, Abe R, Kikuchi K, Hamasaka A, Shinkuma S, Ujiie H, Nomura T, Nishie W, Arita K, Shimizu H. Intraepidermal neutrophilic IgA pemphigus successfully treated with dapsone. European journal of dermatology : EJD. 2012 Mar-Apr:22(2):282-3. doi: 10.1684/ejd.2012.1662. Epub [PubMed PMID: 22378059]
Level 3 (low-level) evidenceLluch-Galcerá JJ, Alcoverro C, Prat E, Martinez-Molina M, Bielsa I, Quer A, Bassas J. [Refraktärer IgA-Pemphigus erfolgreich mit Adalimumab als Monotherapie behandelt]. Journal der Deutschen Dermatologischen Gesellschaft = Journal of the German Society of Dermatology : JDDG. 2023 Dec:21(12):1560-1562. doi: 10.1111/ddg.15232_g. Epub [PubMed PMID: 38082513]
Howell SM, Bessinger GT, Altman CE, Belnap CM. Rapid response of IgA pemphigus of the subcorneal pustular dermatosis subtype to treatment with adalimumab and mycophenolate mofetil. Journal of the American Academy of Dermatology. 2005 Sep:53(3):541-3 [PubMed PMID: 16112379]
Level 3 (low-level) evidenceMoreno AC, Santi CG, Gabbi TV, Aoki V, Hashimoto T, Maruta CW. IgA pemphigus: case series with emphasis on therapeutic response. Journal of the American Academy of Dermatology. 2014 Jan:70(1):200-1. doi: 10.1016/j.jaad.2013.09.037. Epub [PubMed PMID: 24355273]
Level 3 (low-level) evidenceKaegi C, Wuest B, Schreiner J, Steiner UC, Vultaggio A, Matucci A, Crowley C, Boyman O. Systematic Review of Safety and Efficacy of Rituximab in Treating Immune-Mediated Disorders. Frontiers in immunology. 2019:10():1990. doi: 10.3389/fimmu.2019.01990. Epub 2019 Sep 6 [PubMed PMID: 31555262]
Level 1 (high-level) evidenceWatts PJ, Khachemoune A. Subcorneal Pustular Dermatosis: A Review of 30 Years of Progress. American journal of clinical dermatology. 2016 Dec:17(6):653-671 [PubMed PMID: 27349653]
Chatterjee M, Meru S, Vasudevan B, Deb P, Moorchung N. Pemphigus foliaceus masquerading as IgA pemphigus and responding to dapsone. Indian journal of dermatology. 2012 Nov:57(6):495-7. doi: 10.4103/0019-5154.103074. Epub [PubMed PMID: 23248372]
Koga H, Tsutsumi M, Teye K, Ishii N, Yamaguchi M, Nagafuji K, Nakama T. Subcorneal pustular dermatosis-type IgA pemphigus associated with multiple myeloma: A case report and literature review. The Journal of dermatology. 2023 Feb:50(2):234-238. doi: 10.1111/1346-8138.16516. Epub 2022 Jul 15 [PubMed PMID: 35838241]
Level 3 (low-level) evidenceZhou Y, Xiao Y, Wang Y, Li W. Refractory atypical IgA pemphigus successfully treated with apremilast. The Journal of dermatology. 2024 Mar:51(3):e86-e87. doi: 10.1111/1346-8138.17007. Epub 2023 Oct 21 [PubMed PMID: 37864455]