Introduction
Poikilocytosis is the term used for abnormal-shaped red blood cells (RBCs) in the blood. Normal RBCs (also called erythrocytes) are typically disk-shaped, are thinner in the middle than in the edges, with a diameter of 6.2 to 8.2 micrometers, a thickness at the thickest point of 2 to 2.5 micrometers, and a thickness in the center of 0.8 to 1 micrometers. Poikilocytosis generally refers to an increase in abnormal-shaped red blood cells that make up 10% or more of the total red blood cells. Poikilocytes may be flat, elongated, teardrop, or crescent-shaped, or they may have point-like or thorn-like projections or may have any other abnormal feature. Their presence may provide a hematologic 'fingerprint' for identifying their underlying disease pathology.
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
Poikilocytosis is the result of internal damage or external (environmental) trauma. Poikilocytosis causes, generally speaking, it can be inherited or acquired. Inherited conditions can be caused by genetic abnormalities or mutations. Acquired conditions usually develop later in life. The following list provides a general overlay based on etiology.
Inherited causes of poikilocytosis include:
- In sickle cell anemia, RBCs are an abnormal crescent, an elongated spiculated shape known as sickle cells or drepanocytes[1]
- Thalassemia, a genetic disorder in which abnormal hemoglobin is made, contains target cells or codocytes
- Hereditary spherocytosis contains spherocytes[2]
- Pyruvate kinase deficiency contains echinocytes or burr cells
- Hereditary elliptocytosis contains elliptocytes[3]
- McLeod syndrome is a rare genetic disorder that contains acanthocytes
Acquired causes of poikilocytosis include:
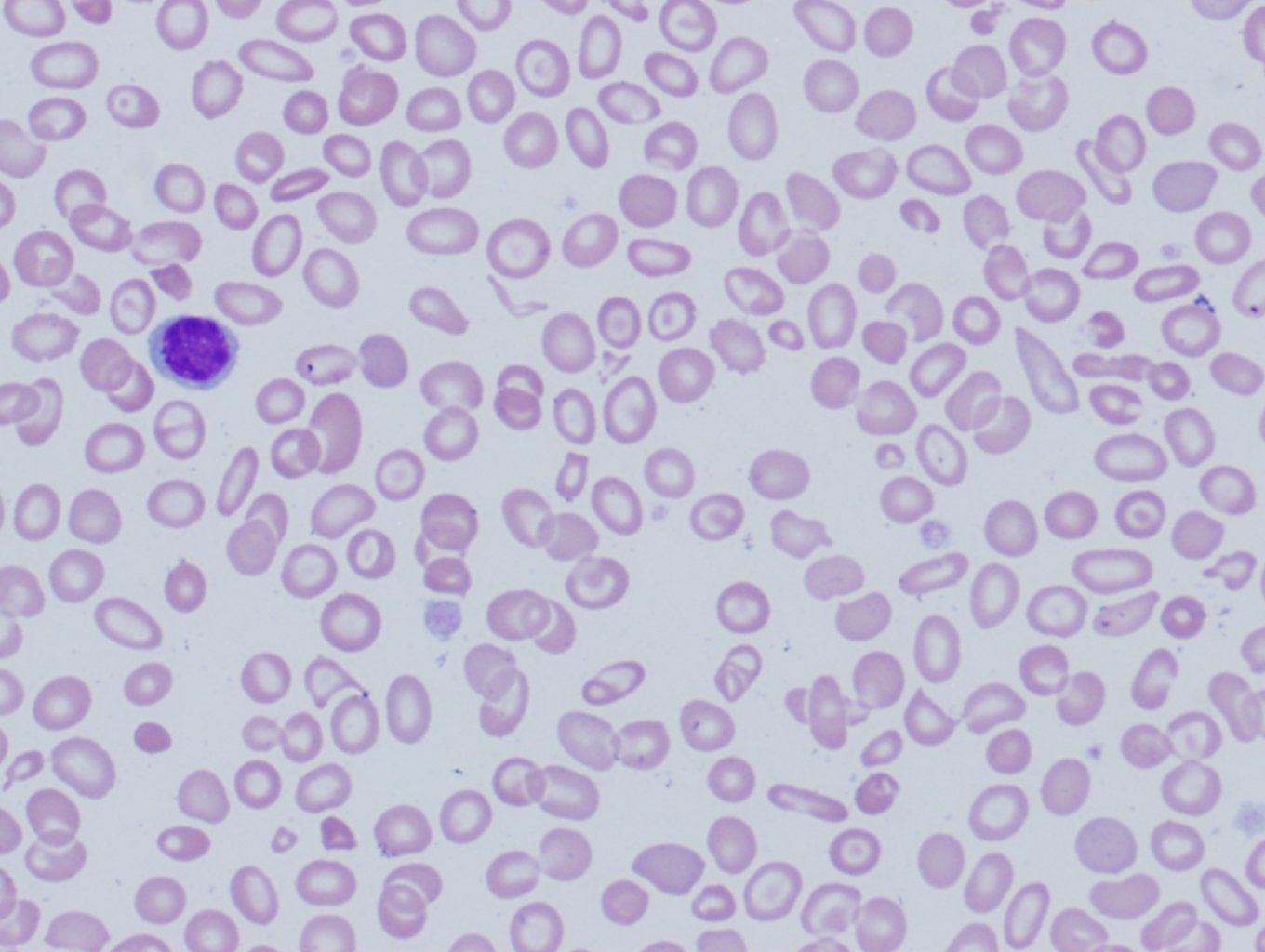
- Iron deficiency anemia, a common type of anemia that is seen when there is insufficient iron in the body, contains elliptocytes (ovalocytes)
- Megaloblastic anemia caused by a deficiency of either folate or vitamin B-12 contains dacrocytes (teardrop cells) and elliptocytes[4][5]
- Autoimmune hemolytic anemia usually contains schistocytes and spherocytes
- Liver and kidney disease contain echinocytes or burr cells
- Alcohol-related liver disease contains acanthocytes
- Myelofibrosis contains tear drop cells
- Lead poisoning (heavy metals)
- Infections (Plasmodium, Babesia)
Causes based on the type of poikilocytosis:
- Spherocytes: Seen in hereditary spherocytosis, autoimmune hemolytic disorders, red cell fragmentation disorders, hemolytic transfusion reactions, ABO hemolytic diseases of newborn/Rh hemolytic disease of the newborn, hypophosphatemia, bartonellosis, snakebites, hyposplenism.
- Codocytes (target cells): Seen in thalassemia, liver disease (cholestatic type), hemoglobin C disorder, recently splenectomy, autosplenectomy in sickle cell disease, and iron deficiency disorder.
- Sickle cells (drepanocytes): Seen in sickle cell disease.
- Dacrocytes (teardrop cells): Seen in beta-thalassemia, leukemia, hemolytic anemia, myelofibrosis, and megaloblastic anemia.
- Acanthocytes (spur cells): Seen in liver disease, thalassemia, abetalipoproteinemia, splenectomized patients, autoimmune hemolytic anemia, kidney disease, and Mcleod syndrome.[6]
- Schistocytes: Seen in hemolytic anemias, sepsis, burns, disseminated intravascular coagulation, platelet disorders like thrombotic thrombocytopenic purpura, and hemolytic uremic syndrome.
- Elliptocytes(ovalocytes): Seen in iron deficiency anemia, thalassemia, megaloblastic anemia, and myelofibrosis.
- Echinocytes (burr cells): Seen in pyruvate kinase deficiency, kidney disease, and cancer.
- Other nonspecific types are leukocytes, stomatocytes (mouth-shaped cells), and degmacytes (bite cells).
Epidemiology
Epidemiology depends on the cause of poikilocytosis. In the case of sickle cell disease, it is autosomal recessive in inheritance and has many variants. Heterozygotes are prevalent in Africa, Meditteranean, the middle east, and India. Thalassemias are of two types: alpha and beta; alpha thalassemia is common in sub-Saharan Africa, the Mediterranean, and generally tropical regions. Beta thalassemia is common in the Mediterranean.[7] Hereditary spherocytosis is common in European and North American ancestry.[2][8] Pyruvate kinase deficiency is most commonly seen in northern Europe and Japan.
Pathophysiology
Pathophysiology varies depending on the cause.[9] Erythrocytes are affected by internal damage or external trauma; with the resultant distortion, the dysmorphism, they may lose pliability, thereby becoming sequestered and either disfigured or destroyed by the spleen.
- Drepanocytes or sickle cells: Seen is sickle cell disease due to hemoglobin S (HbS) by substituting valine in place of glutamic acid at position 6 of beta-globin of HbA. It has an autosomal recessive inheritance. There are many variants like hemoglobin SC disease, hemoglobin S beta thalassemia disease, etc.[10]
- Spherocytes: Seen most commonly in hereditary spherocytosis due to mutations in genes that code for the red blood cell membrane proteins spectrin (alpha and beta), ankyrin, band 3 protein, protein 4.2, and other red blood cell membrane proteins that cause erythrocytes to change shape.[3] Disease entities manifesting spherocytes include hereditary spherocytosis (AKA Minkowski-Chauffard disease), ABO incompatibility, autoimmune hemolytic anemia (warm Antibody subtype), Cold agglutinin hemolysis, severer burns, myelodysplasia (MDS), hemolytic disease of the newborn, and myelophthisis.
- Target cells (codocytes): Most commonly seen in thalassemia due to impaired hemoglobin production (Hb). Normal Hb has two alpha and two beta chains, a decrease in alpha chains is alpha-thalassemia, and a decrease in beta chains is beta-thalassemia.[11] Target cells have an area of central hemoglobinization surrounded by a halo of pallor. Besides hemoglobinopathies, these cells are also found in lecithin-cholesterol acyl transferase (LCAT) deficiency, iron deficiency anemia, sideroblastic anemia, obstructive intrahepatic disease, and status post splenectomy (or asplenia).
- Schistocytes are fragmented RBCs seen most commonly in hemolytic anemias, hemolytic disorders, DIC, pre-eclampsia, B12 deficiency, HIV, BMT, mechanical heart valves, and COVID infection, thrombotic thrombocytopenic purpura and hemolytic uremic syndrome.[12][13][14]
- Echinocytes or Burr cells can be found in a myriad of clinical situations, including burns, uremia, malnutrition, hepatic cirrhosis, pyruvate kinase deficiency, hypomagnesemia, hypophosphatemia, hemodialysis, parenteral fish oil and sometimes in long-distance runners.[15][16][17]
- Acanthocytes, or spur cells, are found in disease entities like myelodysplasia, pyruvate kinase deficiency, alcoholic cirrhosis, hypothyroidism, anorexia nervose, drug-induced forms (e.g., statins, misoprostol), neuroacanthocytosis, and abetalipoproteinemia[6][18][19][20][21] The latter two comprise hereditary neurodegenerative diseases with acanthocytes as a concurrent finding.
- Dacrocytes or teardrop cells / lacrymocytes are erythrocytes that have been remodeled into a 'teardrop formation.'[9][22] While they are highly suggestive of myelofibrosis, they do appear in other ailments. These include thalassemia major, myeolphtisis (marrow infiltration), extramedullary hematopoiesis, hereditary elliptocytosis, hereditary pyropokilocytosis, severe Iron deficiency, myelodysplasia, megaloblastic anemia, autoimmune hemolytic anemia, and microangiopathic hemolytic anemia.
- Elliptocytes or ovalocytes are best exemplified by hereditary elliptocytosis (>25% of cases) and hereditary pyropoikilocytosis. They are both autosomal, the former dominant while the latter is recessive. Other entities that can present with elliptocytes include iron deficiency anemia, thalassemia, myelofibrosis, and myelodysplasia.[23][24][25][26]
- Degmacytes or Bite cells derive their name, colloquially, from the fact that their physical structure gives the impression of a "bite" mark in the membrane. The "bite" occurs as the spleen removes damaged hemoglobin from the cell. This represents a common endpoint to a variety of causations. Diseases involved with this finding include G6PD deficiency, unstable hemoglobins, (congenital) Heinz body anemia, fava beans, and drug-induced oxidative hemolysis.[27][28]
- Stomatocytes are derived from genetic defects in membrane proteins leading to the "leakiness" of cations and fluid.[29] Cells appear cup-shaped with a central pallor in the form of a slit, sometimes referred to as a "fish mouth." The hereditary form of Stomatocytosis is autosomal dominant. Acquired forms include factors such as excessive alcohol intake (that resolves within two weeks of alcohol cessation), obstructive lung disease, Rh-null (autosomal recessive defect in phospholipids), and artifactually (due to slow drying of the specimen in a humid environment.[30]
Histopathology
Drepanocytes; Hemolysis of the sickle cell compounds the illness by potentiating the systemic inflammatory environment.[31][32] With hemolysis, there is a release of cell-free hemoglobin, which depletes Hemopexin, Haptoglobin, and, most notably, nitric oxide.[33] With nitric oxide depletion comes platelet activation, upregulation of endothelial adhesion molecules, and vasoconstriction. These signs herald the development of vascular dysfunction, occlusion, and subsequent end-organ damage. In drepanocytes, the amino acid malposition leads to red cell hypoxic sensitivity and sickling. As the erythrocytes pass through the relatively hypoxic regions of the body's capillaries, the low oxygen tension causes the Hemoglobin S to polymerize into rods leading to the sickle shape primarily and a cigar-shape secondarily.[34][35]
S-polymer damages the cell membranes leading to a calcium/potassium transit problem. The S-polymers also precipitate against the inner surface of the red cell membrane leading to iron-mediated oxidative damage. The damaged sickle cells then adhere to the vascular endothelium leading to vascular occlusion.[31]
Spherocytes; Spherocytes are small round cells that lack the flat, light-colored center of regular erythrocytes. The hereditary form is autosomal dominant in roughly 75% of cases, while the remaining 25% are autosomal recessive or de novo.[36] The hereditary forms have gene mutations SPTA1, SPTB, ANK1, SLC4A1, and EPB42 that code for proteins spectrin, ankyrin, band 3, and erythrocyte protein band 4.2. The resultant dysmorphisms arise from their decreased membrane surface area with decreased membrane flexibility and increased fragility. The cells become rounded and lose their central pallor. They become more easily captured and prematurely destroyed by the spleen. Sometimes the hemolysis may be significant in that the bone marrow initiates a massive (reactive) production of young red blood cells, called reticulocytosis.
Normally reticulocytes are macrocytic and carry a bluish hue due to the presence of internal RNA. They are pitted and matured within the spleen and should normally be < 2.5% of the red cell population. The difference in color tone between the bluish reticulocytes and the red cells is referred to as polychromasia. Autoimmune hemolysis is identified by the presence of a positive Coomb test alongside the spherocytes [36]. Microspherocytes, hyperchromic, and densely hemoglobinized red cells are also found in immune hemolysis.[9] Spherocytes can be acquired in MDS due to the mutation in U2AF1.[37] Spherocytes may be found alongside a myriad of other cell forms in a finding known as leukoerythroblastosis. This tends to occur when the bone marrow is infiltrated or overrun by another disease entity, such as cancer. Typically leukoerythroblastosis portends a poor prognosis.
Codocytes; The basic mechanism of target cell formation is when the intracellular volume has become disproportionately smaller than what is needed to keep the surface area full.[38] The resultant membrane appears redundant, can fold over itself (making a bell-like form), and remodel in such a fashion as to possess a pale halo twixt a red peripheral ring and central core. In liver disease, the membrane cholesterol is decreased, thereby decreasing the membrane tensile strength and leading to codocyte formation. In hemoglobinopathies, there can be uneven hemoglobin distribution within the cell leading to a distortion in the surface area/volume ratio with subsequent target cell formation. Target cells may also occur artifactually when blood smears are made in a high-humidity environment. The margin is up to 5%
Schistocytes; Schistocytes are disrupted red blood cells. Helmet cells are a subset where the red cell has been "chopped" mechanically, leaving behind a roughly semicircular residual. It looks akin to a helmet with a straight border next to the semicircle and sharp, angular edges. Triangular cells are the remnants, the triangle-shaped detritus of red cells; they are very small fragments with significant distortions from significant disruptions. Schistocytes might actually be 'normal' in adults and full-term neonates if their presence is < 1% of the population.[13] In newborns, one may find pyknocytes, fragile and contracted erythrocytes. They can persist in premature babies for up to the first six months of life. Sometimes schistocytes can be difficult to identify if they are embedded in a clouded field of other cell types like echinocytes, acanthocytes, and created (shriveled) red blood cells. In this situation, noting an increase in the LDH, the RDW (measurement of anisocytosis), or the presence of polychromasia may offer a measure of help in making the diagnosis.
Echinocytes or Burr cells are typically reversible dysmorphisms. They have an area of central pallor and multiple small projections, almost spicular, with uniform size and presentation.[6]
Acanthocytes are spiculated erythrocytes with projections of varying size, shape, and distribution.[20] Present theory suggests that expanding the outer red cell membrane layer gives rise to the characteristic spicules. (Expansion of the inner lipid bilayer is thought to lead to stomatocytosis.) Autophagia and autolysosomal degeneration occur in later stages.
Tear Drop Cells; It is believed that the approach to teardrop cell (dacrocyte) formation may be multifocal though the underlying mechanism may be common.[9][22] It is felt that the cells may be disrupted or distorted as they pass through the bone marrow or the splenic sinusoids. Credence is given to this belief in that, in some cases, dacrocytes have disappeared after splenectomy.
Elliptocytes; The basic pathomechanism involved in elliptocytosis involves defects in the horizontal cytoskeletal network of the red cell. This involves problems with the spectrin dimer interaction or the spectrin-actin-protein 4.1 junctional complexes.[39][40] In hereditary elliptocytosis, there are defects in one of the following genes; EPB4I (1p33-p32) erythrocyte membrane protein band 4.1, SPTA1(1q21) alpha=spectrin erythrocyte, SPTB (14q24.1-q24.2) beta-spectrin erythrocyte, and GYPC(2q14-q21) glycophorin C / Gerbich blood groups. Hereditary pyropoikilocytosis is a severe form of elliptocytosis with hemolytic anemia and jaundice in infancy.[40] The cells resemble those from thermal burn cases with microspherocytes and erythrocyte fragments.
Bite cells; The commonality amongst diseases with bite cells (degmacytes) is the denaturation and precipitation of their hemoglobin.[27][41] Once the hemoglobin precipitates, it forms a Heinz body, which will cause the entrapment of the red cells within the spleen. The spleen severs off the affected membrane-hemoglobin segment leaving behind the classic "bite." G6PD deficiency is an x-linked recessive disease prevalent among African and Mediterranean peoples. The enzyme protects the red cell from oxidative stress, but as the cells age, the concentration lessens, and hemolysis can result. A patient with hemolysis may give an impression of normalcy as the body's reaction to the hemolysis is reticulocytosis. Younger red cells have an adequate enzyme amount and so the level, overall, when tested, may appear to be normal. Drugs such as phenazopyridine, nitrofurantoin, and sulfa agents can cause oxidative stress in these cells.[28] Infections and Fava beans are causative agents as well.
Stomatocytes; The basic defect in stomatocytosis is believed to be the dysregulation of fluid and cation movement into the cells. PIEZO1 codes for a transmembrane protein that is felt to establish "mechanosensitive currents" across the red cell membrane.[42][43] PIEZO1 mediates calcium movement and acts as a primary regulator of volume.[30] Variations in the red cell presentations are explained by differences in PIEZO1 gene mutation prevalence.[29] Dysfunction typically leads to dehydration (rarely overhydration).
History and Physical
In poikilocytosis, RBCs are irregularly shaped and may be unable to carry enough oxygen. Subsequently, the patient will manifest fatigue, pale skin, shortness of breath, palpitations, and weakness. The main feature of poikilocytosis is having 10 percent or more abnormally-shaped RBCs. External stressors have the propensity to magnify the signs and symptoms of the erythrocyte's illness. For example, sickle cell disease commonly presents as (when exposed to cold, dehydration, hypoxia) sickle cell crisis (hemolytic, aplastic, sequestration), acute chest syndrome, vaso-occlusive crisis, and autosplenectomy.
Anemia due to increased intravascular hemolysis can present as an indirect hyperbilirubinemia. The sclera and skin may appear jaundiced in hemolysis; the urine may be discolored. The most difficult part of the exam is that the symptoms can be admixed with other, nonspecific findings.
Evaluation
Poikilocytosis is diagnosed with a blood smear examination.[44] This test can be done as part of a routine physical exam or if the patient is experiencing symptoms of anemia or any unexplained symptoms. As stated before, the most difficult part of the evaluation is that the symptoms may be admixed with other, nonspecific findings.
Not all RBCs will take on an abnormal shape. Patients with poikilocytosis have some normally shaped cells RBCs mixed with abnormally shaped poikilocytes. Sometimes, many different types of poikilocytes are present in one patient's blood smear in conditions like iron deficiency anemia, megaloblastic anemia, etc.
In addition to the blood smear, other tests are done to determine the etiology of abnormally shaped RBCs. Examples of other commonly used diagnostic tests include serum iron levels (iron studies), complete blood count (CBC), vitamin B-12, folate, mean corpuscular volume (MCV), mean corpuscular hemoglobin (MCH), mean corpuscular hemoglobin concentration (MCHC), and liver function tests. Choosing the right test(s) can depend on determining the right morphology.
Treatment / Management
The treatment of poikilocytosis is based on the underlying cause of the poikilocytosis. In conditions where poikilocytosis is caused by iron deficiency anemia, megaloblastic anemia due to low levels of iron, vitamin B12, or folate, respectively, is usually treated by taking supplements and increasing the levels of these vitamins in the diet and/or by treating the underlying disease (such as celiac disease or alcoholism) that have caused the deficiency.[45]
Poikilocytocis due to inherited causes like thalassemia or sickle cell anemia may require treatment for a long duration, depending on their clinical variants (homozygous or heterozygous). Genetic diseases, generally, require lifelong monitoring with the treatment of periodic disease exacerbations. These cases may require blood transfusions, or in some cases of poikilocytosis, a bone marrow transplant may be necessary to treat their condition. People with other causes of poikilocytosis, like liver disease (with target cells or acanthocytes), should be treated accordingly. Some may require a liver transplant, while patients with sepsis or serious infections (may have schistocytes) may need antibiotics.
Differential Diagnosis
Poikilocytosis should be differentiated from the following:
- Anisocytosis is the abnormal size of RBCs. As a general rule, hypochromic microcytosis deals with iron deficiencies (with subsequent hemoglobin deficiencies) or hemoglobinopathies, while macrocytic anemia deals with B12 and folate deficiencies or hypothyroidism (causing nuclear/cytoplasmic asynchrony in maturation).[46][47][48] The remaining ailments often fall within the "normochromic normocytic" category.
- Howell Jolly bodies, which are RBCs with inclusions, are seen in conditions like asplenia.
- Anisochromia is variability in the color density of erythrocytes due to unequal hemoglobin, including hypochromic cells seen in iron deficiency anemia.
- Rouleaux are aggregations of RBCs seen when the erythrocyte sedimentation rate is increased in infections, cancers, and inflammatory conditions. Rouleaux formation refers to the stacking of red cells like coins in a single file. It is seen in hypoproteinemia and paraproteinemias like plasma cell disorders as well as in B-cell lymphomas. Rouleaux is differentiated from agglutination, in which there are clusters or clumps of red cells. This is oft an antibody-mediated agglutination and could give a falsely elevated MCV.[49] A false Rouleaux may also be made if the red cell preparation is too thick. The morphology in this situation will be difficult to evaluate because the erythrocytes will be packed.[50]
- Heavy metals (lead); classically, coarse basophilic stippling is associated with heavy metal poisoning (e.g., Pb).[51][52] Basophilic stippling is aggregates of ribosomes and fragments of rRNA precipitates. Patients with hemoglobinopathies also have basophilic stippling, though it is finer in consistency. Chronic lead exposure leads to microcytosis and an increase in the mean platelet volume (MPV) without effect on the platelet count or the leukocyte count (though the differential may be affected).[53] This is felt to be due to heme synthesis interference as well as via effects on hematopoietic cytokines.[54]
Prognosis
The prognosis depends on the cause of poikilocytosis. Some diseases like iron deficiency anemia and megaloblastic anemias have a good prognosis as the disease is resolved by correcting the underlying nutritional defect. However, some causes, like sickle cell disease, may have a poor prognosis depending on presentation and complications, including infections and vaso-occlusive crises, requiring lifelong treatments.
Complications
Complications also depend on the cause of poikilocytosis. Sometimes the severity of the disease may overshadow or obscure the underlying dyserythropoiesis. Clinical acumen, coupled with a thorough observation, can use poikilocytosis to point in the right diagnostic direction. The following are some of the complications associated with common etiologies of poikilocytosis:
Sickle cell disease complications include:
- Infections
- Osteomyelitis
- Stroke, priapism
- Acute chest syndrome
- Hemolytic crisis
- Aplastic crisis
- Autosplenectomy (seen in sickle cell disease)[55]
Hereditary spherocytosis complications include:
- Folate deficiency (might be a functional deficit).
- Hemolysis
- Pigmented gall stones
- Aplastic crisis
Anemia complications include palpitations, heart failure, pregnancy complications, and delayed growth in infants and children.
Deterrence and Patient Education
Poikilocytosis is caused by another medical condition. The patients should be educated about the cause of their poikilocytosis, prognosis, and treatment options. Conditions like anemia caused by iron deficiency are treatable and have a very good prognosis, but they can be dangerous if not managed properly. Anemia during pregnancy can cause complications, including severe congenital defects such as neural tube defects caused by megaloblastic anemia due to folate deficiency. Other complications of anemia during pregnancy include preeclampsia, preterm birth, etc. Anemia caused by a genetic disorder such as sickle cell anemia and thalassemia will require lifelong treatment.
Pearls and Other Issues
- Poikilocytosis is a term for abnormal-shaped red blood cells in the blood.
- Poikilocytosis refers to an increase in abnormal red blood cells of any shape that makes up 10% or more of the total population. Poikilocytes can be flat, elongated, teardrop-shaped, crescent-shaped, sickle-shaped, or can have pointy or thorn-like projections, or may have other abnormal features.
- Poikilocytosis can be due to inherited or acquired causes. Inherited conditions are due to a genetic mutation. Acquired conditions usually develop later in life.
- The most common etiologies of poikilocytosis are sickle cell disease, thalassemia, hereditary spherocytosis, iron deficiency anemia, megaloblastic anemia, and liver disease.
- The most common types of poikilocytosis are sickle cells, target cells, spherocytes, elliptocytes, ovalocytes, echinocytes, and acanthocytes.
- They are usually identified on the blood smear and play an important role in diagnosing the underlying cause and its management.
- Treatment and prognosis vary depending on the cause of the poikilocytosis.
Enhancing Healthcare Team Outcomes
The outcome of poikilocytosis depends on the type of poikilocytosis, its cause, and how quickly the underlying disease is diagnosed and managed. It requires a well-planned interprofessional team approach with a primary clinician, a hematologist, a hematopathologist, a pharmacist, and a nurse, all working together as a healthcare team to bring the best outcome to the patients. The patients should be educated about the cause, prognosis, and different treatment options available. It is essential to identify and diagnose the condition early. Red blood cell morphology can be likened to a trail, a track, from which, with other current data, the proper diagnosis can be pursued and found.
Media
(Click Image to Enlarge)
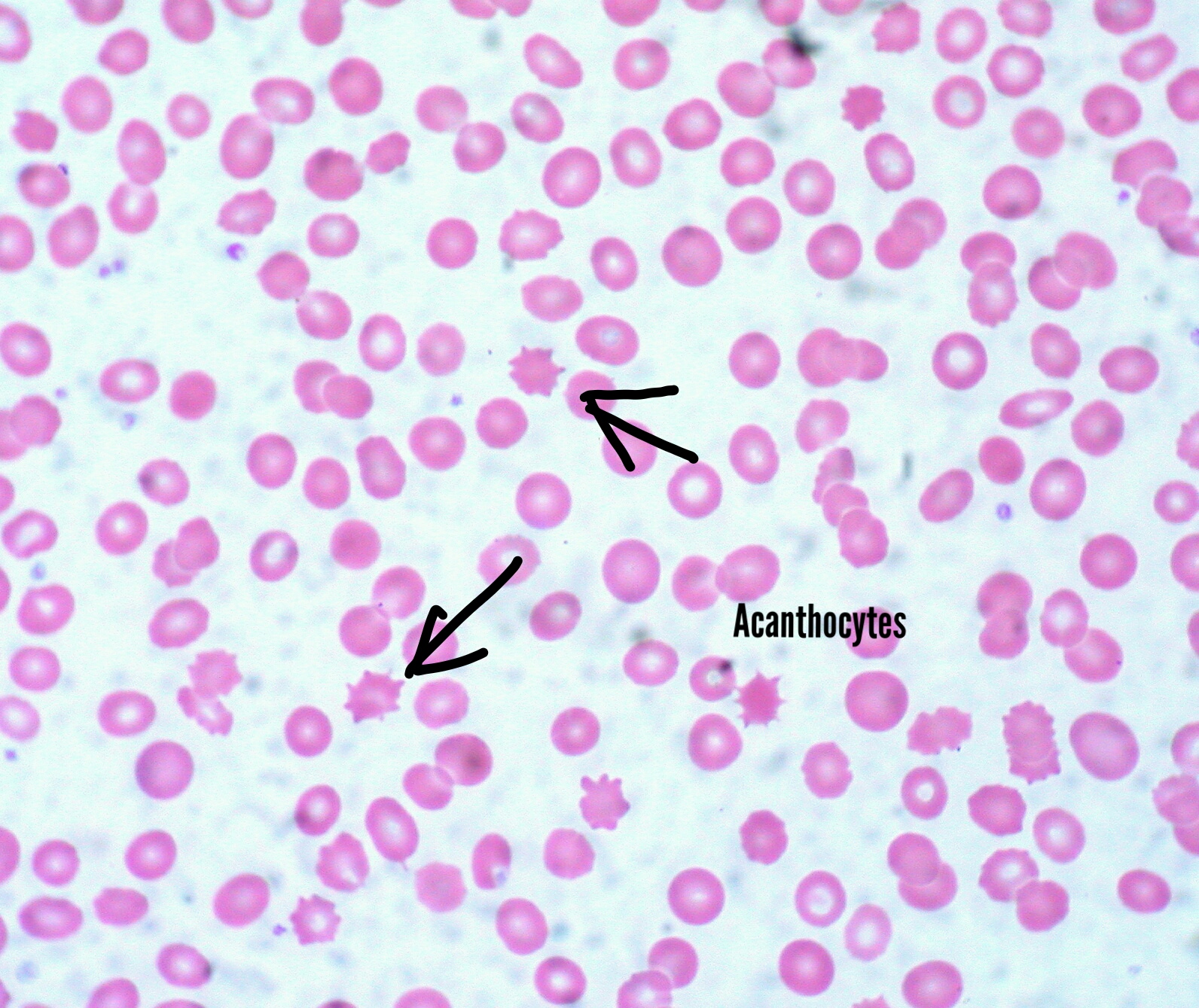
Acanthocytes found in the peripheral blood smear of abetalolipoproteinemia patients. Contributed by Ed Uthman
(Click Image to Enlarge)
Peripheral blood with features of beta-thalassemia minor. Microcytosis and frequent target cells are characteristic. Contributed by David T Lynch, MD
(Click Image to Enlarge)
Iron deficiency anemia Image courtesy S Bhimji MD
(Click Image to Enlarge)
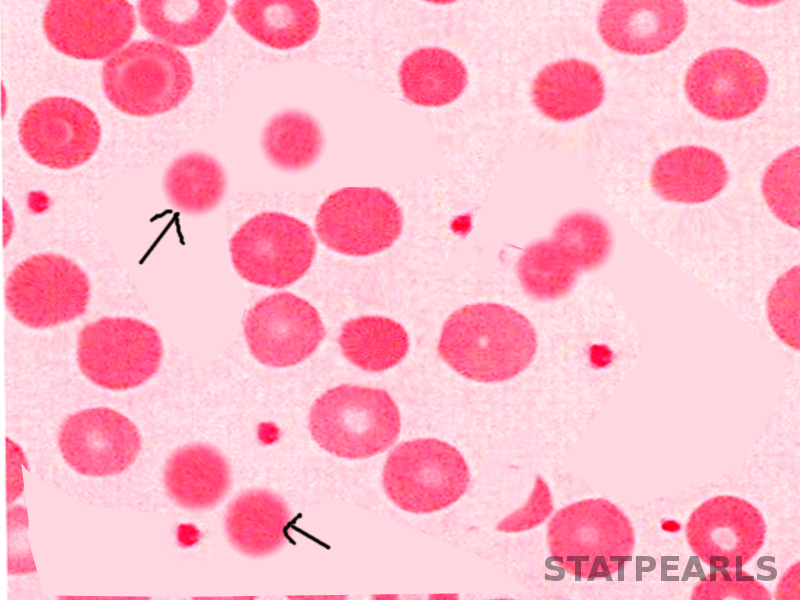
Hereditary Spherocytosis Image courtesy S Bhimji MD
(Click Image to Enlarge)
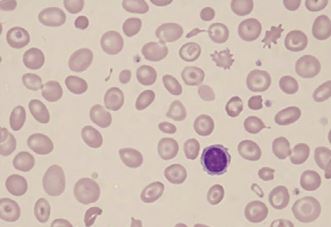
Peripheral blood picture of beta thalassemia major patient showing hypochromic, microcytic red blood cells along with target cells. Contributed by Hamza Bajwa, MD
References
Mangla A, Ehsan M, Agarwal N, Maruvada S. Sickle Cell Anemia. StatPearls. 2023 Jan:(): [PubMed PMID: 29489205]
Zamora EA, Schaefer CA. Hereditary Spherocytosis. StatPearls. 2023 Jan:(): [PubMed PMID: 30969619]
Narla J, Mohandas N. Red cell membrane disorders. International journal of laboratory hematology. 2017 May:39 Suppl 1():47-52. doi: 10.1111/ijlh.12657. Epub [PubMed PMID: 28447420]
Wickramasinghe SN. Diagnosis of megaloblastic anaemias. Blood reviews. 2006 Nov:20(6):299-318 [PubMed PMID: 16716475]
Green R, Datta Mitra A. Megaloblastic Anemias: Nutritional and Other Causes. The Medical clinics of North America. 2017 Mar:101(2):297-317. doi: 10.1016/j.mcna.2016.09.013. Epub 2016 Dec 14 [PubMed PMID: 28189172]
Shah PR, Grewal US, Hamad H. Acanthocytosis. StatPearls. 2023 Jan:(): [PubMed PMID: 31747195]
Martin A, Thompson AA. Thalassemias. Pediatric clinics of North America. 2013 Dec:60(6):1383-91. doi: 10.1016/j.pcl.2013.08.008. Epub 2013 Oct 4 [PubMed PMID: 24237977]
Manciu S, Matei E, Trandafir B. Hereditary Spherocytosis - Diagnosis, Surgical Treatment and Outcomes. A Literature Review. Chirurgia (Bucharest, Romania : 1990). 2017 Mar-Apr:112(2):110-116. doi: 10.21614/chirurgia.112.2.110. Epub [PubMed PMID: 28463670]
Adewoyin AS, Nwogoh B. Peripheral blood film - a review. Annals of Ibadan postgraduate medicine. 2014 Dec:12(2):71-9 [PubMed PMID: 25960697]
Sedrak A, Kondamudi NP. Sickle Cell Disease. StatPearls. 2024 Jan:(): [PubMed PMID: 29494006]
Olivieri NF. The beta-thalassemias. The New England journal of medicine. 1999 Jul 8:341(2):99-109 [PubMed PMID: 10395635]
Usuki K. [Anemia: From Basic Knowledge to Up-to-Date Treatment. Topic: IV. Hemolytic anemia: Diagnosis and treatment]. Nihon Naika Gakkai zasshi. The Journal of the Japanese Society of Internal Medicine. 2015 Jul 10:104(7):1389-96 [PubMed PMID: 26513958]
Zini G, d'Onofrio G, Erber WN, Lee SH, Nagai Y, Basak GW, Lesesve JF, International Council for Standardization in Hematology (ICSH). 2021 update of the 2012 ICSH Recommendations for identification, diagnostic value, and quantitation of schistocytes: Impact and revisions. International journal of laboratory hematology. 2021 Dec:43(6):1264-1271. doi: 10.1111/ijlh.13682. Epub 2021 Aug 24 [PubMed PMID: 34431220]
Desai K, Sridhar A, Matos J, Mulla S, Thirumaran R. Thrombotic Thrombocytopenia Masquerading As COVID-19 Infection. Cureus. 2022 Jul:14(7):e26933. doi: 10.7759/cureus.26933. Epub 2022 Jul 17 [PubMed PMID: 35989804]
Hasler CR, Owen GR, Brunner W, Reinhart WH. Echinocytosis induced by haemodialysis. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association. 1998 Dec:13(12):3132-7 [PubMed PMID: 9870478]
Koralkova P, van Solinge WW, van Wijk R. Rare hereditary red blood cell enzymopathies associated with hemolytic anemia - pathophysiology, clinical aspects, and laboratory diagnosis. International journal of laboratory hematology. 2014 Jun:36(3):388-97. doi: 10.1111/ijlh.12223. Epub [PubMed PMID: 24750686]
Mallah HS, Brown MR, Rossi TM, Block RC. Parenteral fish oil-associated burr cell anemia. The Journal of pediatrics. 2010 Feb:156(2):324-6.e1. doi: 10.1016/j.jpeds.2009.07.062. Epub [PubMed PMID: 20105643]
Level 3 (low-level) evidenceHuang S, Zhang J, Tao M, Lv Y, Xu L, Liang Z. Two case reports of chorea-acanthocytosis and review of literature. European journal of medical research. 2022 Feb 7:27(1):22. doi: 10.1186/s40001-022-00646-7. Epub 2022 Feb 7 [PubMed PMID: 35130982]
Level 3 (low-level) evidencePeikert K, Hermann A, Danek A. XK-Associated McLeod Syndrome: Nonhematological Manifestations and Relation to VPS13A Disease. Transfusion medicine and hemotherapy : offizielles Organ der Deutschen Gesellschaft fur Transfusionsmedizin und Immunhamatologie. 2022 Feb:49(1):4-12. doi: 10.1159/000521417. Epub 2022 Jan 25 [PubMed PMID: 35221863]
Yu Y, Lu Y, Wang F, Lu Y, Xie B, Meng X, Tang Y. Acanthocytes Identified in Huntington's Disease. Frontiers in neuroscience. 2022:16():913401. doi: 10.3389/fnins.2022.913401. Epub 2022 Jun 6 [PubMed PMID: 35733931]
Cloos AS, Daenen LGM, Maja M, Stommen A, Vanderroost J, Van Der Smissen P, Rab M, Westerink J, Mignolet E, Larondelle Y, Terrasi R, Muccioli GG, Dumitru AC, Alsteens D, van Wijk R, Tyteca D. Impaired Cytoskeletal and Membrane Biophysical Properties of Acanthocytes in Hypobetalipoproteinemia - A Case Study. Frontiers in physiology. 2021:12():638027. doi: 10.3389/fphys.2021.638027. Epub 2021 Feb 23 [PubMed PMID: 33708142]
Level 3 (low-level) evidenceRobier C, Klescher D, Reicht G, Amouzadeh-Ghadikolai O, Quehenberger F, Neubauer M. Dacryocytes are a common morphologic feature of autoimmune and microangiopathic haemolytic anaemia. Clinical chemistry and laboratory medicine. 2015 Jun:53(7):1073-6. doi: 10.1515/cclm-2014-0936. Epub [PubMed PMID: 25503671]
Kjelland JD, Dwyre DM, Jonas BA. Acquired Elliptocytosis as a Manifestation of Myelodysplastic Syndrome with Ring Sideroblasts and Multilineage Dysplasia. Case reports in hematology. 2017:2017():3625946. doi: 10.1155/2017/3625946. Epub 2017 Oct 11 [PubMed PMID: 29158926]
Level 3 (low-level) evidenceQuigley M, Linfesty RL, Bethel K, Sharpe R. Stubby elliptocytes are an invariable feature of leukoerythroblastosis. Blood. 2007 Mar 15:109(6):2666 [PubMed PMID: 17341668]
Level 3 (low-level) evidenceJiménez Gonzalo FJ, de Luis Navarro J, de Blas Orlando JM, Martín Noya A. [Hereditary elliptocytosis associated with heterozygous beta-thalassemia with a hemolytic component]. Sangre. 1999 Oct:44(5):391-2 [PubMed PMID: 10618922]
Level 3 (low-level) evidenceRodgers MS, Chang CC, Kass L. Elliptocytes and tailed poikilocytes correlate with severity of iron-deficiency anemia. American journal of clinical pathology. 1999 May:111(5):672-5 [PubMed PMID: 10230358]
Kachur ME, Rosen BJ. Educational Case: Anemia in a Neonate. Academic pathology. 2021 Jan-Dec:8():23742895211002829. doi: 10.1177/23742895211002829. Epub 2021 Apr 8 [PubMed PMID: 33889717]
Level 3 (low-level) evidencePrchal JT, Gregg XT. Red cell enzymes. Hematology. American Society of Hematology. Education Program. 2005:():19-23 [PubMed PMID: 16304354]
Lee AC, Aung L, Yip YY, Hia CP. Hereditary stomatocytosis: an unusual cause of severe neonatal jaundice. Singapore medical journal. 2018 Sep:59(9):505. doi: 10.11622/smedj.2018115. Epub [PubMed PMID: 30310921]
Flatt JF, Bruce LJ. The Molecular Basis for Altered Cation Permeability in Hereditary Stomatocytic Human Red Blood Cells. Frontiers in physiology. 2018:9():367. doi: 10.3389/fphys.2018.00367. Epub 2018 Apr 16 [PubMed PMID: 29713289]
Moerdler S, Manwani D. New insights into the pathophysiology and development of novel therapies for sickle cell disease. Hematology. American Society of Hematology. Education Program. 2018 Nov 30:2018(1):493-506. doi: 10.1182/asheducation-2018.1.493. Epub [PubMed PMID: 30504350]
Salinas Cisneros G, Thein SL. Research in Sickle Cell Disease: From Bedside to Bench to Bedside. HemaSphere. 2021 Jun:5(6):e584. doi: 10.1097/HS9.0000000000000584. Epub 2021 Jun 1 [PubMed PMID: 34095767]
Mack AK, Kato GJ. Sickle cell disease and nitric oxide: a paradigm shift? The international journal of biochemistry & cell biology. 2006:38(8):1237-43 [PubMed PMID: 16517208]
Kavanagh PL, Fasipe TA, Wun T. Sickle Cell Disease: A Review. JAMA. 2022 Jul 5:328(1):57-68. doi: 10.1001/jama.2022.10233. Epub [PubMed PMID: 35788790]
Eaton WA, Bunn HF. Treating sickle cell disease by targeting HbS polymerization. Blood. 2017 May 18:129(20):2719-2726. doi: 10.1182/blood-2017-02-765891. Epub 2017 Apr 6 [PubMed PMID: 28385699]
Wu Y, Liao L, Lin F. The diagnostic protocol for hereditary spherocytosis-2021 update. Journal of clinical laboratory analysis. 2021 Dec:35(12):e24034. doi: 10.1002/jcla.24034. Epub 2021 Oct 24 [PubMed PMID: 34689357]
Karlsson LK, Mottelson MN, Helby J, Petersen J, Glenthøj A. Acquired spherocytosis in the setting of myelodysplasia. Leukemia research reports. 2022:17():100332. doi: 10.1016/j.lrr.2022.100332. Epub 2022 Jun 2 [PubMed PMID: 35720514]
CROSBY WH. The pathogenesis of spherocytes and leptocytes (target cells). Blood. 1952 Feb:7(2):261-74 [PubMed PMID: 14886421]
Anil More T, Kedar P. Unravelling the genetic and phenotypic heterogeneity of SPTA1 gene variants in Hereditary Elliptocytosis and Hereditary Pyropoikilocytosis patients using next-generation sequencing. Gene. 2022 Nov 15:843():146796. doi: 10.1016/j.gene.2022.146796. Epub 2022 Aug 9 [PubMed PMID: 35961434]
Bayhan T, Ünal Ş, Gümrük F. Hereditary Elliptocytosis with Pyropoikilocytosis. Turkish journal of haematology : official journal of Turkish Society of Haematology. 2016 Mar 5:33(1):86-7. doi: 10.4274/tjh.2015.0054. Epub 2015 Aug 6 [PubMed PMID: 26377499]
Opsahl M, Chen W. Blister and bite cells in G6PD deficiency. International journal of laboratory hematology. 2022 Feb:44(1):55-56. doi: 10.1111/ijlh.13689. Epub 2021 Aug 24 [PubMed PMID: 34431226]
Andolfo I, Alper SL, De Franceschi L, Auriemma C, Russo R, De Falco L, Vallefuoco F, Esposito MR, Vandorpe DH, Shmukler BE, Narayan R, Montanaro D, D'Armiento M, Vetro A, Limongelli I, Zuffardi O, Glader BE, Schrier SL, Brugnara C, Stewart GW, Delaunay J, Iolascon A. Multiple clinical forms of dehydrated hereditary stomatocytosis arise from mutations in PIEZO1. Blood. 2013 May 9:121(19):3925-35, S1-12. doi: 10.1182/blood-2013-02-482489. Epub 2013 Mar 11 [PubMed PMID: 23479567]
Level 3 (low-level) evidenceGallagher PG. Disorders of red cell volume regulation. Current opinion in hematology. 2013 May:20(3):201-7. doi: 10.1097/MOH.0b013e32835f6870. Epub [PubMed PMID: 23519154]
Level 3 (low-level) evidenceRashid A. A 65-year-old man with anemia: diagnosis with peripheral blood smear. Blood research. 2015 Sep:50(3):129. doi: 10.5045/br.2015.50.3.129. Epub 2015 Sep 22 [PubMed PMID: 26457277]
DeLoughery TG. Iron Deficiency Anemia. The Medical clinics of North America. 2017 Mar:101(2):319-332. doi: 10.1016/j.mcna.2016.09.004. Epub 2016 Dec 8 [PubMed PMID: 28189173]
Kumar SB, Arnipalli SR, Mehta P, Carrau S, Ziouzenkova O. Iron Deficiency Anemia: Efficacy and Limitations of Nutritional and Comprehensive Mitigation Strategies. Nutrients. 2022 Jul 20:14(14):. doi: 10.3390/nu14142976. Epub 2022 Jul 20 [PubMed PMID: 35889932]
Kohne E. Hemoglobinopathies: clinical manifestations, diagnosis, and treatment. Deutsches Arzteblatt international. 2011 Aug:108(31-32):532-40. doi: 10.3238/arztebl.2011.0532. Epub 2011 Aug 8 [PubMed PMID: 21886666]
Level 2 (mid-level) evidenceAslinia F, Mazza JJ, Yale SH. Megaloblastic anemia and other causes of macrocytosis. Clinical medicine & research. 2006 Sep:4(3):236-41 [PubMed PMID: 16988104]
Waider KL. Rouleaux and saline replacement. Immunohematology. 2018 Sep:34(3):91-92 [PubMed PMID: 30295502]
Abramson N. Rouleaux formation. Blood. 2006 Jun 1:107(11):4205 [PubMed PMID: 16739263]
Level 3 (low-level) evidenceZamani A, Kazerooni ES, Kasaee SS, Anbardar MH, Mohammadzadeh S, Shekarkhar G, Soleimani N. And the Oscar Goes to Peripheral Blood Film for the Detection of Lead Poisoning in a Complicated Toxic Patient: A Case Report with a Review of Laboratory Clues. Case reports in medicine. 2022:2022():9238544. doi: 10.1155/2022/9238544. Epub 2022 Feb 23 [PubMed PMID: 35251184]
Level 3 (low-level) evidenceKano N, Fukui S, Kushiro S, Inui A, Saita M, Kura Y, Sawada U, Naito T. Basophilic stippling in red blood cells in the bone marrow: indication for lead poisoning diagnosis. The Journal of international medical research. 2022 Feb:50(2):3000605221078405. doi: 10.1177/03000605221078405. Epub [PubMed PMID: 35184610]
Chwalba A, Maksym B, Dobrakowski M, Kasperczyk S, Pawlas N, Birkner E, Kasperczyk A. The effect of occupational chronic lead exposure on the complete blood count and the levels of selected hematopoietic cytokines. Toxicology and applied pharmacology. 2018 Sep 15:355():174-179. doi: 10.1016/j.taap.2018.05.034. Epub 2018 May 29 [PubMed PMID: 29857081]
Dobrakowski M, Boroń M, Czuba ZP, Birkner E, Chwalba A, Hudziec E, Kasperczyk S. Blood morphology and the levels of selected cytokines related to hematopoiesis in occupational short-term exposure to lead. Toxicology and applied pharmacology. 2016 Aug 15:305():111-117. doi: 10.1016/j.taap.2016.06.015. Epub 2016 Jun 11 [PubMed PMID: 27298078]
Porter M. Rapid Fire: Sickle Cell Disease. Emergency medicine clinics of North America. 2018 Aug:36(3):567-576. doi: 10.1016/j.emc.2018.04.002. Epub [PubMed PMID: 30037443]