Introduction
The initiation and coordination of movement are under the control of an immense network of nerves in the central nervous system (CNS) that originate from the cerebral cortex and course through the internal capsule, brainstem, and spinal cord. The impulses for movement are carried by nerves known as upper motor neurons (UMN). The pyramidal tract is the primary tract that propagates signals necessary for voluntary movement. The pyramidal tract divides into the corticospinal tract and the corticobulbar tract. Injury to UMNs in these tracts is common because of the large areas covered by the motor neuron pathway. Any injury to these tracts is known as UMN lesions. Damage to UMNs results in characteristic clinical manifestations colloquially termed “upper motor neuron signs” or “upper motor neuron syndrome.” The symptoms include muscle weakness, spasticity, hyperreflexia, and clonus. Damage to UMNs of the corticobulbar tract can manifest as dysphagia and dysarthria. Distinguishing upper motor neuron signs from lower motor neuron signs is essential in the neurological physical exam.[1]
Structure and Function
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Structure and Function
Upper motor neuron signs are secondary to damage to upper motor neurons. UMNs are first-order neurons that carry the electrical impulses for movement, and many descending UMN tracts coordinate movement. The pyramidal tract is the principal UMN tract that initiates voluntary movement. This tract provides a direct path between the cerebral cortex and spinal cord. Lesions at any point along the pyramidal tract in the brain or spinal cord can manifest as UMN signs.[2]
The pyramidal tract divides into the corticospinal and corticobulbar tracts. Pyramidal tract cell bodies concentrate primarily in the motor area of the cerebral cortex. Motor areas organize somatotopically. The motor areas of the left and right hemispheres innervate musculature on the contralateral side of the body. For example, an upper motor neuron lesion in the left motor area will manifest UMN signs on the right side of the body.[2]
From the cell bodies in the motor areas of the cerebral cortex, UMN axons travel through the following ipsilateral structures in sequential order: corona radiata, posterior limb of the internal capsule, cerebral peduncle of the midbrain, central pons, and pyramids of the medulla. At the pyramids, corticospinal tract axons crossover at the pyramidal decussation. The axons are now contralateral from their cell bodies of origin, and they enter the spinal cord at the lateral funiculus. In the spinal cord, the tract is the lateral corticospinal tract. Axons terminate at various levels of the spinal cord within the central gray column and base of the dorsal column. They can either terminate on interneurons or directly on lower motor neurons. UMNs that synapse directly onto lower motor neurons typically control more distal extremities. These direct connections are thought to be necessary for the fine control of the fingers and hands.[3]
Corticobulbar tract axons follow a similar trajectory to the corticospinal tract, traveling through the corona radiata and internal capsule. Once the tract reaches the level of the brainstem, the axons synapse onto cranial nerve nuclei at the appropriate level. Most cranial nerves receive bilateral innervation from UMNs in the left and right hemispheres. Exceptions to this rule include parts of CN VII and XII. These cranial nerves only receive contralateral innervation from the pyramidal tract.[3]
The lateralization of UMN signs is dependent on the location of UMN lesions. Lesions above the pyramidal decussation manifest as symptoms contralateral to the lesion. UMN lesions below the decussation cause symptoms ipsilateral to the site of the lesion. Unilateral lesions of UMNs to most cranial nerves tract do not manifest with significant symptoms because of their bilateral innervation from the motor areas with the exceptions of CN VII and XII.[4]
Clinical Significance
Damage to descending motor pathways in the CNS results in UMN neuron signs. This can be due to various pathologies such as trauma, cerebrovascular accidents, infections, malignancies, neurodegenerative disorders, and metabolic disorders. It is important to distinguish upper motor neuron signs from lower motor neuron signs during the physical exam. Lower motor neuron signs typically present with muscle atrophy, paralysis of individual muscles, fasciculations, fibrillations, hypotonia, and hyporeflexia. UMN signs can be divided into negative and positive signs. Positive signs refer to symptoms that exhibit increased muscle activity and exaggerated spinal reflexes. These include hyperreflexia, propagation of reflexes, clonus, spasticity, flexor and extensor spasms, co-contraction, synkinesias, and spastic dystonia. Negative symptoms are those that involve loss of motor control. These include weakness, loss of dexterity, fatigue, and impaired motor planning and control. Alpha motor neurons are preserved in UMN lesions, therefore muscle atrophy, fasciculations, and fibrillation are not present as UMN signs. The specific constellation of UMN signs is dependent on the location of UMN lesions. The onset of these UMN signs is preceded by a period of spinal shock following a UMN lesion. Understanding the range of clinical symptoms is important in the assessment of weakness.[5][6]
Spinal Shock
Spinal shock typically refers to the interval of acute flaccid paralysis following spinal cord lesions. UMN lesions that are secondary to spinal cord damage result in flaccid paralysis of voluntary movement, lack of motor response to external stimuli, and the abolishment of spinal reflexes below the level of the lesion. This presentation of areflexia and hypotonia can also be seen in cerebral UMN lesions; however, it is not as prominent as spinal cord lesions. Generally, the more severe the UMN lesion, the more severe the symptoms. The motor findings in spinal shock are often accompanied by sensory or autonomic abnormalities depending on the specific spinal lesion. These symptoms include paralysis of the bowels and bladder, loss of sensation below corresponding spinal levels, and loss of vasomotor tone below the level of the lesion. Spinal shock is believed to be caused by the deprivation of spinal circuits of input from descending motor pathways from the cerebellum and brainstem. In a span of a few days to weeks, the spinal cord gradually regains its function, and the symptoms of spasticity and hyperreflexia develop. The exact mechanism for this transition of symptoms is still unknown.[7]
Weakness
A defining characteristic of upper motor neuron weakness is its tendency to affect muscle groups rather than individual muscles as in lower motor neuron lesions. In addition, the paralysis induced by UMN lesions varies based on the severity of the damage. This is in contrast to the complete paralysis seen in lower motor neurons lesion due to the destruction of the alpha motor neurons targeting specific muscles. In the face, there is a characteristic weakness of contralateral lower facial muscles due to UMN lesions at the cerebral, midbrain, and pontine levels. The weakness of the extremities is most prominent on distal musculature compared to proximal muscles. In addition, fine finger movements are most affected due to the loss of upper motor neurons that directly innervate alpha motor neurons. Finally, there is a distinct pattern of weakness observed where the antigravity muscles of the body are most noticeably affected. For example, the extensors of the upper arm are weaker than the flexors whereas the flexors of the leg are weaker than the extensors. Other patterns of weakness in the upper extremity include weakness of wrist extension compared to flexion and weakness of shoulder abduction compared to adduction. Patterns of weakness in the lower extremity include weakness of hip abduction compared to the adduction and weakness of ankle eversion compared to inversion.[8][9]
The pronator drift test is an indicator of UMN weakness. The test is performed by asking the patient to hold their arms outstretched in front of them with the palms face up, fingers spread wide, and eyes closed. The clinician may observe the hands beginning to close, pronation of the arms, and downward drift of the arms. The findings of this test are due to the weakness of the supinator compared to the pronator muscles of the arm.[9]
Spasticity
Spasticity is clinically defined as a velocity-dependent increase in muscle tone. This presents as increased resistance to passive lengthening. The mechanism is related to the disinhibition of spinal reflex arcs causing increased excitability of muscle stretch reflex. As previously stated, spasticity follows a period of spinal shock due to the adaptation of spinal neuronal pathways. On physical exam, slow extension or flexion of the arms or legs will not elicit increased muscle tone. In contrast, performing brisk stretches of the of muscles elicits abrupt increases in tone followed by decreased muscular resistance. This constitutes the clasp knife phenomenon of spasticity. In addition to the velocity dependence of spasticity, it is also a length-dependent movement. There has been no linear association seen between the spasticity and weakness observed with UMN lesions. Some patients with severe weakness only exhibit mild spasticity while severe cases of spasticity are associated with minimal levels of muscle weakness. This suggests that the mechanisms for weakness and spasticity follow different paths from the central nervous system.[10]
Hyperreflexia
The loss of inhibitory activity from descending motor results in disinhibition of spinal reflex circuits. This, in turn, leads to exaggerated deep tendon reflexes such as the knee jerk reflex on the physical exam. Radiation of reflexes is a commonly observed sign in a patient in the spastic state. For example, tapping on the radial periosteum may result in reflex contraction of the biceps, triceps, and finger flexors in addition to the brachioradialis. This is most likely due to the radiation of vibration waves from bone to muscle.[11]
Clonus
Hyper-reflexive signs can take the form of rhythmic, involuntary contractions known as clonus. The contractions occur at frequencies between 5 and 7 Hz and are most commonly seen at the ankle and patella. It is performed by rapidly stretching and sustaining a stretch at a constant length around a muscle joint.[12]
Babinski and other reflexes
Damage to the descending motor pathways causes loss of modulation of spinal reflexes. One of the most reliable upper motor neuron signs is the Babinski reflex. This is performed by stroking the lateral sole of a foot from the heel to the toe with a firm but painless stimulus. A positive sign is seen with an extension of the large toe along with extension and fanning of the remaining toes. The Babinski reflex is commonly seen in neonates but diminishes as the descending motor pathways mature. The reemergence of the reflex is strongly indicative of damage to the pyramidal tract. There are a number of Babinski-like reflexes that elicit an abnormal plantar response. Examples of these include the Chaddock sign, Moniz sign, and Oppenheim sign. The Chaddock sign is elicited by stroking the lateral malleolus. Moniz sign is tested by forcefully and passively plantar flexing the ankle. Finally, the Oppenheim sign is examined by placing pressure on the medial side of the tibia. A testable upper extremity reflex is the Hoffman reflex. The reflex is performed by stabilizing a patient’s middle finger and quickly flicking the tip of the finger. The reflex is positive if the fingers and thumb flex.[13][14]
Hyporeflexia of superficial reflexes
The superficial reflexes are motor reactions in response to light stimulation of overlying skin. Classic superficial reflexes are the abdominal reflex, cremasteric reflex, and the corneal reflex. Decreased intensity of the superficial reflexes can be a sign of upper motor neuron damage. However, they may be difficult to analyze because the reflexes may be absent in normal individuals while it may reemerge in patients with UMN lesions. The mechanism of the hyporeflexia is not known.[15]
Synkinesias
Synkinesias are the involuntary movement of one limb following the voluntary movement of a different limb. For example, voluntary flexion of the arm may result in flexion of the leg or dorsiflexion of the foot. These involuntary movements may also occur with certain automatisms such as yawning and sneezing.[6]
Co-contraction
Physiologic co-contraction is the concomitant contraction of agonist and antagonist muscle groups in preparation for a movement or as a response to environmental stimuli to maintain adequate tension around a joint. A pathologic co-contraction as a UMN sign is a hyperactive stretch reflex in an antagonist muscle generated in response to normal contraction of an agonistic muscle, which results in decreased rates of rapid alternating movement. There is ultimately greater fatigability and decreased ability to perform voluntary movements.[16]
Pseudobulbar Palsy and Cranial Nerves VII and XII
Nearly all cranial nerves receive bilateral innervation from the corticobulbar tract. Exceptions to this are cranial nerves VII and XII. Parts of CN VII and XII only receive unilateral innervation from higher-order processing centers. Because of the bilateral innervation of most cranial nerves, unilateral UMN lesions of the corticobulbar tract will not result in paralysis or weakness. Bilateral lesions to the corticobulbar tract results in a set of clinical findings known as pseudobulbar palsy. Acutely, pseudobulbar palsy has a progression of symptomatology similar to spinal shock. Patients can present obtunded, comatose, or severely demented. Patients will also lose all ability to speak or swallow. As the patient recovers, their symptoms will give way to dysarthria, dysphagia, dysphonia, spastic tongue, pseudobulbar affect, and exaggerated facial reflexes. On facial reflex testing, a brisk jaw jerk can present with normal or increased palatal reflexes. Pseudobulbar affect, also known as emotional incontinence or emotional lability, are episodes of unintentional crying or laughing.[17][18]
Parts of cranial nerves XII and the lower division of cranial nerve VII are unique in their unilateral innervation from higher-order motor centers. Unilateral lesions of UMN supplying these cranial nerves are sufficient in creating clinical deficits. A unilateral lesion to the UMN of CN VII manifests as a contralateral lower facial droop. A lesion to the UMN of XII manifests as tongue deviation pointing away from the side of the lesion.[19][20]
Other Issues
A thorough history and complete physical and neurological exam are essential in ascertaining the etiology of upper motor neuron signs. The differential for UMN signs includes strokes, malignancies, traumatic brain injury, infections, inflammatory diseases, neurodegenerative diseases, and metabolic disorders. The following are select diseases that present with UMN signs.
Strokes are defined as the sudden cessation of blood flow to various areas of the brain resulting in neuronal cell death. Due to the vast areas spanned by the pyramidal tract, upper motor neurons are vulnerable to many stroke syndromes. Strokes subdivide into two categories: ischemic or hemorrhagic. The sudden interruption of blood flow secondary to thrombi, emboli, or compression defines ischemic strokes. Sudden bleeding into the brain secondary to the rupture of a blood vessel defines hemorrhagic strokes. The target area of an upper motor neuron lesion is dependent on the branch of the cerebral blood supply that is damaged. Motor areas of the cerebral cortex are damaged with occlusion of the middle cerebral artery or the anterior cerebral artery. Strokes involving these arteries are also likely to present with sensory, perceptual, visual, and language deficits. Damage to the posterior limb internal capsule secondary to occlusion at the lenticulostriate arteries presents with pure UMN deficits of the contralateral face, arm, and leg.[21]
The pyramidal tract can undergo impingement by mass effect secondary to malignancies or abscesses. The majority of brain malignancies are due to metastasis from another primary tumor. Common malignancies that metastasize to the brain include lung cancer, breast cancer, melanoma, colon cancer, and renal cancer. Treatment for metastatic brain tumors involves a combination of surgery, radiation, and chemotherapy. Infections can localize to specific areas in the brain and create empyemas, abscesses, or cysts. Some organisms that create focal cysts within the brain include Aspergillus, toxoplasma gondi, and taenia solium.[22]
Neurodegenerative diseases are a collection of disorders that primarily affect neurons. The most common neurodegenerative disease is amyotrophic lateral sclerosis (ALS). ALS is unique in its involvement of upper and lower motor neurons. The clinical symptoms are a mix of upper and lower motor neuron signs. A diagnosis is made with nerve conduction studies and electromyography. Labs are ordered to rule out differential diagnoses that also manifest with muscle weakness. ALS is currently incurable.[23]
Autoimmune diseases can damage neurons disrupting their ability to conduct signals from the brain. One example is multiple sclerosis (MS) which is an immune-mediated, inflammatory, demyelinating disease. It is characterized by symptoms that are separated by time and space. Presentation of multiple sclerosis is highly variable and can include UMN signs, cognitive disturbance, visual changes, ataxia, and sensory changes. The imaging test of choice to diagnose MS is MRI. Intrathecal immunoglobulin G and oligoclonal bands are common findings in a cerebrospinal fluid analysis of an MS patient that can support the diagnosis.[24]
Spinal cord lesions that damage the corticospinal tract can present with UMN signs. Brown-Sequard syndrome is a lesion due to hemisection of the spinal cord. The most common etiology is penetrating trauma to the spine. Other causes include blunt trauma, hematoma, tumors, and disc herniation. UMN signs manifest ipsilateral and below the level of the lesion. Patients will also present with signs of damage to the dorsal column and lateral spinothalamic tract; this presents as ipsilateral loss of fine touch, vibration, proprioception, and contralateral loss of pain and temperature sensation.[25]
Treatment
Treatment of UMN signs starts with the diagnosis of the underlying cause of the symptoms. Appropriate medical or surgical management is then pursued based on the underlying disease process. However, in cases where there is no immediate cure, management of the long term sequelae of upper motor neuron lesions is essential. Spasticity management is a major issue that requires attention due to the profound disability incurred by patients. It can lead to a restriction in the activity, pain, weakness, and contractures. Optimal management requires an understanding of the pathophysiology and natural history of spasticity. The aims of spasticity management include improvement of function, symptom relief, and improvement of posture.[26]
The management of spasticity involves physical and medical modalities. Physical rehabilitation is a mainstay in treatment and involves proper positioning of patients, stretching of muscles, cooling of muscles, heat, and orthosis. Pharmacologic management should be an adjunct to these physical modalities. Correct positioning is an important area of management, especially for immobilized patients. The main goal is to produce a stretch of spastic muscles to reduce spasticity and facilitate the function of antagonistic muscles. Proper stretching maintains muscle length throughout passive and active exercises. Stretching plans can include standing and splinting for short and long term management. Application of heat can increase the elasticity and thus relax spastic muscles; this should be utilized in conjunction with stretching and exercise. Finally, orthosis can be used to distribute or remove forces from the body to allow better control of movement or alter body shape. Orthoses can include ankle supports, insoles, wrist splints, knee splints, spinal braces, or neck collars. Splints and casts can be used to prevent the formation of contractures in the long term.[26]
Medical management aims to improve function and relieve symptoms and is always in conjunction with physical modalities. Medications can be administered enterally, intrathecally, and intramuscularly. Nerve blocks are also an option in certain cases. Common oral medications include baclofen, dantrolene, benzodiazepines, and gabapentin. Oral medications are typically for milder cases of spasticity. The dosages required to provide relief in severe cases often present with unwanted side effects such as drowsiness and weakness. Nerve blocks are another avenue of treatment that can be of use in severe cases of spasticity. A nerve block is the application of a chemical agent that impairs the conduction along a nerve. Common agents utilized are phenol, alcohol, and local anesthetics. The major indication for a nerve block is debilitating and painful spasticity. Nerve blocks can be performed with fluoroscopy or nerve stimulation. To test the effects of a nerve block on a muscle, peripheral blocks with local anesthetics can be used to observe potential benefits and side effects.[27]
Media
(Click Image to Enlarge)
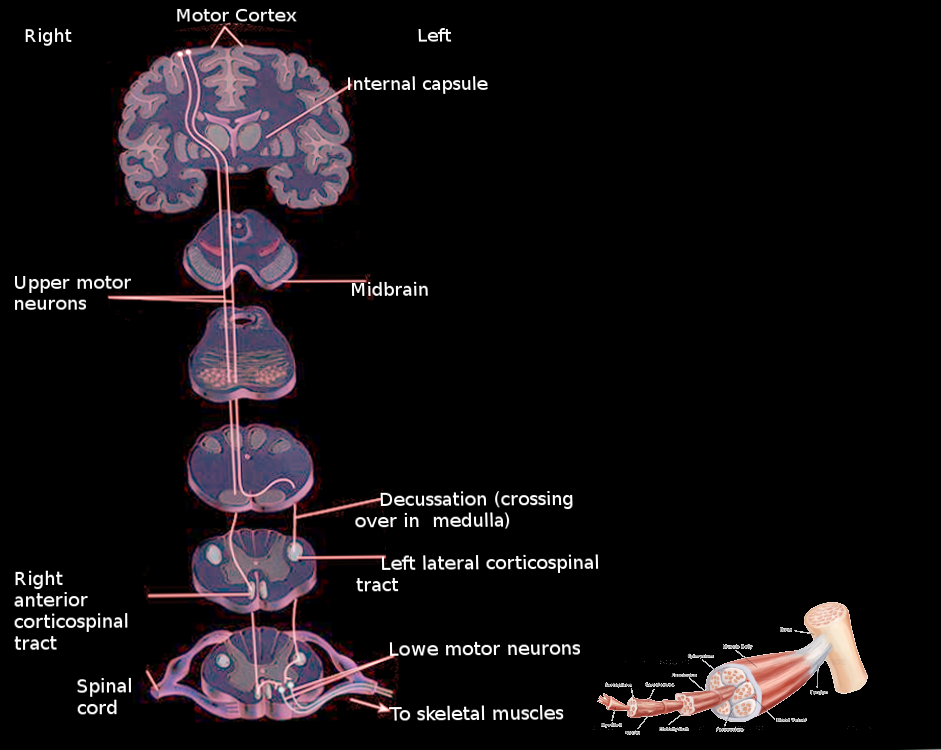
Pyramidal tract Image courtesy Dr Chaigasame
References
Emos MC, Agarwal S. Neuroanatomy, Upper Motor Neuron Lesion. StatPearls. 2023 Jan:(): [PubMed PMID: 30725990]
Jang SH. The corticospinal tract from the viewpoint of brain rehabilitation. Journal of rehabilitation medicine. 2014 Mar:46(3):193-9. doi: 10.2340/16501977-1782. Epub [PubMed PMID: 24531325]
Canedo A. [Functional heterogeneity of the piramidal system: corticobulbar and corticospinal tracts]. Revista de neurologia. 2003 Mar 1-15:36(5):438-52 [PubMed PMID: 12640598]
Level 3 (low-level) evidenceArmand J. [The pyramidal tract. Recent anatomic and physiologic findings]. Revue neurologique. 1984:140(5):309-29 [PubMed PMID: 6379818]
Level 3 (low-level) evidenceMayer NH, Esquenazi A. Muscle overactivity and movement dysfunction in the upper motoneuron syndrome. Physical medicine and rehabilitation clinics of North America. 2003 Nov:14(4):855-83, vii-viii [PubMed PMID: 14580042]
Mayer NH, Herman RM. Phenomenology of muscle overactivity in the upper motor neuron syndrome. Europa medicophysica. 2004 Jun:40(2):85-110 [PubMed PMID: 16046932]
Atkinson PP, Atkinson JL. Spinal shock. Mayo Clinic proceedings. 1996 Apr:71(4):384-9 [PubMed PMID: 8637263]
Colebatch JG, Gandevia SC. The distribution of muscular weakness in upper motor neuron lesions affecting the arm. Brain : a journal of neurology. 1989 Jun:112 ( Pt 3)():749-63 [PubMed PMID: 2731028]
Adams RW, Gandevia SC, Skuse NF. The distribution of muscle weakness in upper motoneuron lesions affecting the lower limb. Brain : a journal of neurology. 1990 Oct:113 ( Pt 5)():1459-76 [PubMed PMID: 2245306]
Mukherjee A, Chakravarty A. Spasticity mechanisms - for the clinician. Frontiers in neurology. 2010:1():149. doi: 10.3389/fneur.2010.00149. Epub 2010 Dec 17 [PubMed PMID: 21206767]
LANCE JW, DEGAIL P. SPREAD OF PHASIC MUSCLE REFLEXES IN NORMAL AND SPASTIC SUBJECTS. Journal of neurology, neurosurgery, and psychiatry. 1965 Aug:28(4):328-34 [PubMed PMID: 14338123]
Dimitrijevic MR, Nathan PW, Sherwood AM. Clonus: the role of central mechanisms. Journal of neurology, neurosurgery, and psychiatry. 1980 Apr:43(4):321-32 [PubMed PMID: 6445412]
Walker HK, Hall WD, Hurst JW, Walker HK. The Plantar Reflex. Clinical Methods: The History, Physical, and Laboratory Examinations. 1990:(): [PubMed PMID: 21250238]
van Gijn J. The Babinski reflex. Postgraduate medical journal. 1995 Nov:71(841):645-8 [PubMed PMID: 7494766]
Dick JP. The deep tendon and the abdominal reflexes. Journal of neurology, neurosurgery, and psychiatry. 2003 Feb:74(2):150-3 [PubMed PMID: 12531937]
Heitmann S, Ferns N, Breakspear M. Muscle co-contraction modulates damping and joint stability in a three-link biomechanical limb. Frontiers in neurorobotics. 2011:5():5. doi: 10.3389/fnbot.2011.00005. Epub 2012 Jan 11 [PubMed PMID: 22275897]
Ahmed A, Simmons Z. Pseudobulbar affect: prevalence and management. Therapeutics and clinical risk management. 2013:9():483-9. doi: 10.2147/TCRM.S53906. Epub 2013 Nov 29 [PubMed PMID: 24348042]
Kumar AS, Subrahmanyam DK. Neurocysticercosis presenting as pseudobulbar palsy. Journal of neurosciences in rural practice. 2014 Jan:5(1):76-7. doi: 10.4103/0976-3147.127883. Epub [PubMed PMID: 24741260]
Level 3 (low-level) evidenceSanders RD. The Trigeminal (V) and Facial (VII) Cranial Nerves: Head and Face Sensation and Movement. Psychiatry (Edgmont (Pa. : Township)). 2010 Jan:7(1):13-6 [PubMed PMID: 20386632]
Walker HK, Hall WD, Hurst JW, Walker HK. Cranial Nerve XII: The Hypoglossal Nerve. Clinical Methods: The History, Physical, and Laboratory Examinations. 1990:(): [PubMed PMID: 21250229]
Lee SJ, Lee DG, Moon HJ, Lee TK. Lesion Pattern, Mechanisms, and Long-Term Prognosis in Patients with Monoparetic Stroke: A Comparison with Nonmonoparetic Stroke. BioMed research international. 2017:2017():9373817. doi: 10.1155/2017/9373817. Epub 2017 Sep 12 [PubMed PMID: 29138753]
Sharma V, Prabhash K, Noronha V, Tandon N, Joshi A. A systematic approach to diagnosis of cystic brain lesions. South Asian journal of cancer. 2013 Apr:2(2):98-101. doi: 10.4103/2278-330X.110509. Epub [PubMed PMID: 24455569]
Level 1 (high-level) evidencede Carvalho M, Poliakov A, Tavares C, Swash M. Interplay of upper and lower motor neuron degeneration in amyotrophic lateral sclerosis. Clinical neurophysiology : official journal of the International Federation of Clinical Neurophysiology. 2017 Nov:128(11):2200-2204. doi: 10.1016/j.clinph.2017.08.024. Epub 2017 Sep 19 [PubMed PMID: 28957737]
Kamińska J, Koper OM, Piechal K, Kemona H. Multiple sclerosis - etiology and diagnostic potential. Postepy higieny i medycyny doswiadczalnej (Online). 2017 Jun 30:71(0):551-563 [PubMed PMID: 28665284]
Zeng Y, Ren H, Wan J, Lu J, Zhong F, Deng S. Cervical disc herniation causing Brown-Sequard syndrome: Case report and review of literature (CARE-compliant). Medicine. 2018 Sep:97(37):e12377. doi: 10.1097/MD.0000000000012377. Epub [PubMed PMID: 30213001]
Level 3 (low-level) evidenceGhai A, Garg N, Hooda S, Gupta T. Spasticity - Pathogenesis, prevention and treatment strategies. Saudi journal of anaesthesia. 2013 Oct:7(4):453-60. doi: 10.4103/1658-354X.121087. Epub [PubMed PMID: 24348300]
Viel E, Pellas F, Ripart J, Pélissier J, Eledjam JJ. [Peripheral nerve blocks and spasticity. Why and how should we use regional blocks?]. Presse medicale (Paris, France : 1983). 2008 Dec:37(12):1793-801. doi: 10.1016/j.lpm.2008.07.007. Epub 2008 Sep 4 [PubMed PMID: 18775634]