Introduction
Alpha-1 antitrypsin (AAT) deficiency is a genetically inherited disorder often unrecognized in clinical practice. It results in impaired production of alpha-1 antitrypsin protein, which plays a role in protecting the body from neutrophil elastase, an enzyme released by white blood cells during infection. Due to defective protein production, there is reduced activity of AAT in the blood and lungs. Additionally, abnormal AAT levels can lead to the accumulation of AAT in the liver, leading to liver disease.
Mutations in the SERPINA1 gene are responsible for both deficiency and the presence of abnormal AAT protein. Inadequate levels of functional AAT result in the destruction of alveoli in the lungs by neutrophil elastase, leading to lung disease. Abnormal AAT can also accumulate in the liver and cause damage to this organ.
Individuals with AAT deficiency usually experience the initial manifestations of lung disease, such as emphysema and bronchiectasis, between the age range of 20 to 50 years. Extensive research has consistently shown that cigarette smoking plays a significant role in lung function decline.[1][2][3] This activity will review the etiology, epidemiology, clinical manifestations, diagnosis, and management of AAT deficiency.
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
AAT Genetics
AAT deficiency is inherited through autosomal co-dominant transmission, which indicates that affected individuals have inherited an abnormal AAT gene from both parents. The gene that encodes AAT is SERPINA1, located on the long arm of chromosome 14.
Over 150 alleles of AAT (SERPINA1) have been identified, and each allele is assigned a letter code based on the protein's electrophoretic mobility that it produces. The most common version (allele) of the SERPINA1 gene is referred to as the "M" allele, which is considered the normal allele. Most individuals in the general population possess two copies of the M allele (MM) in each cell—variations of the SERPINA1 gene result in decreased levels of alpha-1 antitrypsin. For example, the S allele leads to moderately low levels of this protein, while the Z allele results in significantly reduced AAT. Individuals who carry 2 copies of the Z allele (ZZ) in each cell will likely have AAT deficiency. Those with the SZ combination are more likely to develop lung diseases, such as emphysema, particularly if they smoke.
Worldwide, it is estimated that 161 million people carry 1 copy of the S or Z allele and 1 copy of the M allele in each cell (MS or MZ). Individuals with an MS (or SS) combination typically produce sufficient amounts of alpha-1 antitrypsin to protect their lungs. However, people carrying MZ alleles have a slightly increased risk of experiencing impaired lung or liver function.
AAT Phenotypes
AAT phenotypes are based on the electrophoretic mobility of the proteins produced by the various abnormal AAT alleles. Genotyping is performed by identifying specific alleles in DNA.
Based on this, variants of AAT can be categorized into four basic groups:[4]
- Normal is associated with normal levels of AAT and its proper functioning. The group of normal alleles is referred to as M, and the typical genotype with normal AAT levels is MM.
- Deficient is associated with a deficiency in plasma AAT levels below 35% of the average normal level. Homozygotes with AAT deficiency typically have 5 to 6 μmol/L plasma levels. The most common deficient allele associated with emphysema is the Z allele, which is present in approximately 2% to 3% of the Caucasian population in the United States.
- Null alleles result in the absence of detectable AAT protein in the plasma. Therefore, individuals with the null genotype are less common and are at higher risk for the most severe form of lung disease, while liver disease is not typically associated with null alleles.
- Dysfunctional alleles are characterized by producing a normal quantity of AAT protein, but the protein fails to function properly. This dysfunction can be attributed to either a conversion defect from an elastase inhibitor to a thrombin inhibitor or the presence of the PI*F variant.[5][6]
Epidemiology
AAT deficiency is a condition that is present worldwide, but its prevalence varies among different populations. This condition affects approximately 1 in 2,000 to 6,000 individuals and predisposes them to liver disease and early-onset emphysema. AAT deficiency is more prevalent among individuals of European ancestry and relatively less common among individuals of Asian descent and other ancestries.[6][7]
While AAT deficiency is classified as a rare condition, it is believed that approximately 80,000 to 100,000 individuals in the United States have a severe deficiency of AAT, suggesting that the disease is likely under-recognized. Globally, it is estimated that more than 3 million people have allele combinations associated with severe AAT deficiency.[8][9]
Pathophysiology
AAT is a protease inhibitor produced in the liver to protect the lung against damage caused by neutrophil elastase. When AAT misfolds, it forms abnormal polymers that accumulate as hepatocyte inclusions. These inclusions can be detected as positive on periodic acid–Schiff staining with diastase digestion (PAS-D). The presence of misfolded AAT protein can induce hepatotoxic stress, contributing to the development of liver disease. Likewise, AAT deficiency leads to excess neutrophil elastase, which promotes mucin production and enhances the expression of other proteases and inflammatory cytokines.[10]
Environmental factors, such as exposure to tobacco smoke, chemicals, and dust, likely impact the severity of alpha-1 antitrypsin deficiency. The underlying causes of AAT deficiency vary depending on the affected organ. In the case of AAT emphysema, the primary etiology is an imbalance between neutrophil elastase in the lungs and the elastase inhibitor AAT.[10] This imbalance becomes more pronounced when exposed to factors such as cigarette smoke and infections, increasing the elastase burden in the lung, and leading to accelerated lung tissue degradation.[11]
Severe deficiency of AAT is a significant risk factor for early-onset emphysema, although not all individuals with severe deficiency will develop emphysema.[11] Risk factors contributing to emphysema development include cigarette smoking, dusty occupational exposure, parental history of chronic obstructive pulmonary disease (COPD), and a personal history of asthma, chronic bronchitis, or pneumonia.
AAT-related liver cirrhosis occurs due to the accumulation of abnormal AAT protein within the hepatocytes.[12] The liver disease develops due to the accumulation of unsecreted variant AAT protein within the hepatocytes. Only genotypes associated with the pathologic polymerization of AAT within the endoplasmic reticulum of hepatocytes produce the disease. Most patients with AAT-related liver disease are homozygous for the Z allele (PI*ZZ genotype). Liver disease is not observed in null homozygotes with severe AAT deficiency but lack intra-hepatocytic accumulation.
History and Physical
AAT deficiency primarily affects three different organs, leading to distinct clinical manifestations: the lungs, the liver, and, in rare cases, the skin.
The clinical presentation of lung diseases associated with AAT deficiency, such as emphysema, shares many similarities with typical COPD, including a significant portion of affected individuals having a smoking history. However, two distinct characteristics of AAT-related emphysema are the early onset of symptoms at a young age and a basilar-predominant pattern of emphysema.[13] Individuals with AAT-related emphysema experience symptoms at a younger age and typically have a lower forced expiratory volume in 1 second (FEV1) values. According to data from the National Institute of Health-sponsored registry for patients with AAT, the mean FEV1 was 43% ± 30% of predicted, and the mean age was 46 ± 11 years.[14]
Dyspnea is the most common presenting symptom among individuals with AAT deficiency. Many patients also experience a chronic cough, sputum production, and wheezing, both chronically or with upper respiratory tract infections. Spontaneous secondary pneumothorax may be the initial presenting symptom of AAT deficiency or a complication of the underlying disease. Severe AAT deficiency has also been linked to the development of bronchiectasis.
Clinical presentation of extrapulmonary disease in patients with at-risk alleles (eg, Z, S[iiyama], and M[malton]) may develop adult-onset chronic hepatitis, cirrhosis, or hepatocellular carcinoma.
AAT deficiency can also manifest in various extrapulmonary ways. These include necrotizing panniculitis, erythematous nodules, or plaques on the thigh or buttocks, which are the primary dermatologic manifestation of the condition. Other extrapulmonary manifestations are systemic vasculitis (such as Wegner granulomatosis), intracranial and peripheral aneurysms, psoriasis, urticaria, angioedema, potential association with inflammatory bowel disease, fibromuscular dysplasia, peripheral neuropathy, and glomerulonephritis.[15][16][17]
Evaluation
Most individuals affected by AAT deficiency remain undiagnosed due to limited access to adequate healthcare and treatment.[13] Screening for AAT deficiency is essential and should be conducted using various diagnostic methods. It is crucial to maintain a low threshold for clinical suspicion. In addition to testing first-degree relatives of individuals with AAT deficiency, it is important to screen patients with one or more of the following disorders:[10][18]
- COPD: Emphysema type in a young individual (≤45 years), nonsmoker or minimal smoker, or display a predominant basilar radiological pattern.
- Unexplained liver disease or family history of liver disease
- Poorly responsive asthma
- c-ANCA vasculitis (in >90% of cases, the antibody is specific for proteinase 3)
- Panniculitis
- Bronchiectasis not related to cystic fibrosis
Initial evaluation for clinically suspected ATT deficiency includes:
- Medical history & physical examination
- Liver-function tests
- Complete pulmonary function tests (PFT): PFT is used to evaluate the presence and extent of lung disease. This includes spirometry, typically obtained before and after bronchodilator administration, and assessments of lung volumes and diffusing capacity for carbon monoxide (DLCO). A six-minute walk test should be conducted if DLCO falls below the normal range or if the patient reports exertional dyspnea. All adults with persistent airflow obstruction on spirometry, particularly those residing in regions with a high prevalence of AAT deficiency, should undergo testing for AAT deficiency.
- Chest radiography: A chest X-ray is used to determine the pattern and extent of emphysema and to exclude other causes of dyspnea. The “classic” pattern of emphysema observed in AAT deficiency typically manifests as basilar-predominant emphysematous bullae. However, a range of patterns from basilar-predominant to apical-predominant emphysema may be seen. Some clinicians perform chest computed tomography (CT) scans for an initial assessment.[19][20]
- Confirm AAT deficiency: Severe AAT deficiency is diagnosed by confirming a serum level below 11 μmols/L (approximately 57 mg/dL by nephelometry), along with the presence of a severely deficient genotype identified through testing for the most common defective alleles (ie, S, Z, I, F).
If the AAT serum level is greater than 20 μmol/L, the patient is unlikely to have a clinically significant AAT deficiency. However, genotyping becomes necessary if there is a need to assess the presence of particular mutations or identify heterozygotes and mutations with incomplete penetrance.
The normal plasma concentration of AAT ranges from 80 mg/dL to 220 mg/dL (20 to 48 μmol/L using nephelometry or 150 mg/dL to 350 mg/dL by radial immunodiffusion). However, given the variability in reference ranges, patients with a serum AAT level below 100 mg/dL (18.4 μmols/L) should be evaluated further with isoelectric focusing or genotyping.
Isoelectric focusing is the gold standard blood test for identifying AAT variants and is commonly used as a phenotype test.
Genotyping of the protease inhibitor (Pi) locus is performed on a blood sample using polymerase chain reaction (PCR) technology or restriction fragment length polymorphisms. These tests detect the most commonly known variants (F, I, S, Z). In cases where these tests fail to determine the genetic variant, gene sequencing of exonic DNA can be employed as an alternative method.
When monitoring asymptomatic patients, who do not exhibit respiratory symptoms and have normal baseline spirometry (ie, FEV1 80% or greater than predicted), spirometry should be repeated if symptoms change at 6 to 12-month intervals. An unexplained decrease in the post-bronchodilator FEV1 to less than 80% predicted is an indication to consider initiating augmentation therapy.
There is a lack of specific guidelines for monitoring liver disease in patients homozygous for PiZ, PiS[iiyama], or PiM[malton]. It is recommended to assess serum aminotransferases, alkaline phosphatase, and bilirubin annually. Additionally, some clinicians also obtain a complete blood count (CBC) to check for thrombocytopenia and perform abdominal ultrasounds every 6 to 12 months to evaluate for the presence of cirrhosis.
The phenotype presentation of AAT deficiency can vary, even in individuals with confirmed low levels of AAT. The variation is strongly linked to factors such as exposure to cigarette smoking and occupational exposures, including exposure to kerosene or dust.[21]
Treatment / Management
It is crucial for all patients suspected or diagnosed with AAT deficiency to quit smoking. Individuals with severe AAT deficiency, such as those with PI ZZ or null genotypes, should undergo regular monitoring with pulmonary function testing and 6-minute walk testing, clinical assessments, and evaluations of the quality of life. These monitoring activities should be conducted periodically, typically every 6 to 12 months, to assess disease progression and the effectiveness of interventions.[13]
Supportive treatments include inhaled bronchodilators and inhaled or oral glucocorticoids, as recommended by the guidelines for managing COPD.[22] Additional considerations should include evaluating the need for home oxygen supplementation if resting oxygen saturation falls below 88%. Preventive vaccination against respiratory viral infections, such as influenza, and pneumococcal pneumonia, should be prioritized.[23] Pulmonary rehabilitation and nutritional support are also important components of comprehensive care for these patients.[24](A1)
Intravenous augmentation, which involves the infusion of pooled plasma-purified human alpha-1 antitrypsin (an alpha-1 proteinase inhibitor), is the only licensed disease-specific therapy for AAT deficiency associated with lung disease. It was licensed by the Food and Drug Administration in 1987 and has been the most direct and efficient means of elevating AAT levels in the plasma and the lung interstitium.[13][25][26][27] Although augmentation therapy has been shown to decrease the loss of lung density in individuals with AAT deficiency, there is no evidence that it impacts lung function (FEV1), quality of life, or rates of COPD exacerbation.[28]
The American Thoracic Society suggests weekly augmentation therapy with human pooled AAT for individuals with plasma levels of AAT less than 11 μmols/L and established airflow obstruction, defined as an FEV1 less than 80% predicted. On the other hand, the Canadian Thoracic Society suggests keeping AAT augmentation therapy for AAT-deficient patients (AAT level less than 11 μmols/L) with an FEV1 of 25% to 80% predicted who have quit smoking and are on optimal medical therapy. The thoracic society of Australia and New Zealand recommend augmentation therapy to be considered in non-smoking patients with AAT deficiency (as a conditional recommendation and low-quality evidence).[29]
The selection criteria for augmentation therapy include the following:
- High-risk phenotype
- Plasma AAT level below 11 μmols/L
- Airflow obstruction by spirometry (eg, FEV1 <80% of predicted)
- Likely compliance with the protocol
- Age ≥18 years
- Nonsmoker or ex-smoker
Augmentation therapy is not recommended for patients with heterozygous phenotypes, whose plasma AAT level exceeds 11 μmols/L, or current smokers.
Side effects associated with intravenous AAT infusion are uncommon, and no long-term reactions have been noted. However, some side effects can occur, including:
- Low-grade fever and mild flu-like symptoms are usually self-limited.
- Anaphylaxis with IgE antibody formation to AAT has been reported, which is extremely rare.
- A syndrome of transient fever, chest and low back pain, and thrombocytopenia has been reported. This is due to a high molecular weight contaminant in the stabilizer added to the AAT product.
- Pooled human plasma alpha 1-antiprotease contains small amounts of IgA, so IgA-deficient individuals with anti-IgA antibodies are at risk of anaphylaxis with current infusions. Therefore, checking for IgA deficiency ((level <7 mg/dL) or anti-IgA antibodies is recommended before initiating intravenous AAT therapy.
While lung transplantation has been shown to provide both quality of life and survival benefits for selected patients with severe AAT deficiency, other therapies, such as lung volume reduction therapy, are not recommended due to a lack of evidence supporting its use.[29]
Management of extrapulmonary manifestations of AAT deficiency requires the following:
- Regular assessment of liver function is recommended at least annually.[4]
- In contrast to AAT-related lung disease, augmentation therapy is not recommended for isolated AAT-related liver diseases.
- Individuals with AAT deficiency and cirrhosis should undergo screening for hepatocellular carcinoma using liver ultrasound and serum alpha-fetoprotein.[30]
- Liver transplantation is the treatment of choice for AAT deficiency associated with advanced liver disease.
Differential Diagnosis
The differential diagnosis of AAT deficiency lung disease may include the following conditions:
- Emphysema
- Chronic bronchitis
- Bronchiectasis
The differential diagnosis of AAT deficiency liver disease may include the following conditions:
- Chronic viral hepatitis
- Hereditary hemochromatosis
- Wilson disease
- Non-alcoholic steatohepatitis
- Primary biliary cirrhosis
Prognosis
During the first three decades of life, liver dysfunction poses a health threat in individuals with AAT deficiency, while pulmonary dysfunction is less concerning. Beyond the initial decades, the natural history of people with severe AAT deficiency becomes less clear, and survival estimates can vary among studies due to differences in study populations.
Nonsmoking, asymptomatic individuals with AAT deficiency may have relatively normal survival rates, although confirmation through long-term follow-up in a population-based study is necessary. The FEV1 is a major determinant in predicting survival in AAT-deficient individuals, with mortality rising as FEV1 falls below 35% of what is predicted. Other parameters used for prognostic evaluation include decreased lung density as assessed by chest CT.[31][32]
The prognosis for AAT deficiency can vary depending on several factors, including the severity of the disease, the presence of associated complications, and the individual's response to treatment. AAT deficiency-related lung disease can lead to progressive and irreversible lung damage, resulting in COPD, emphysema, and respiratory symptoms.
For individuals with AAT deficiency-related liver disease, the prognosis can also vary. In some cases, liver disease can progress to cirrhosis, which may result in complications such as liver failure or hepatocellular carcinoma. As a result, the prognosis can be grave.
Complications
Complications of alpha-1 antitrypsin deficiency include:
- Asthma
- Emphysema
- Chronic bronchitis
- Chronic liver disease
Deterrence and Patient Education
Patients with alpha-1 antitrypsin deficiency should be advised to quit smoking, avoid exposure to occupational dust, and have yearly influenza and pneumococcal vaccinations. These measures can help prevent the progression of lung disease and reduce the risk of respiratory infections and associated complications.
To prevent secondary complications related to liver disease, it is important to advise patients to limit alcohol intake or abstain from alcohol altogether. Additionally, hepatitis A and B vaccinations should be recommended.
Genetic counseling and testing are recommended for first-degree relatives of individuals with AAT deficiency due to the increased risk of carrying at-risk alleles associated with AAT deficiency.[13][33]
Enhancing Healthcare Team Outcomes
Optimal management of AAT deficiency requires the collaboration of an interprofessional team of healthcare professionals, including a pediatrician, geneticist, pulmonologist, gastroenterologist, and internist. Regular monitoring of lung function, including FEV1, and periodic chest CT scans are essential for assessing disease progression and guiding treatment decisions. Individualized management plans should be developed based on the patient's specific needs, risk factors, and disease progression to preserve lung function and optimize long-term outcomes
Media
(Click Image to Enlarge)
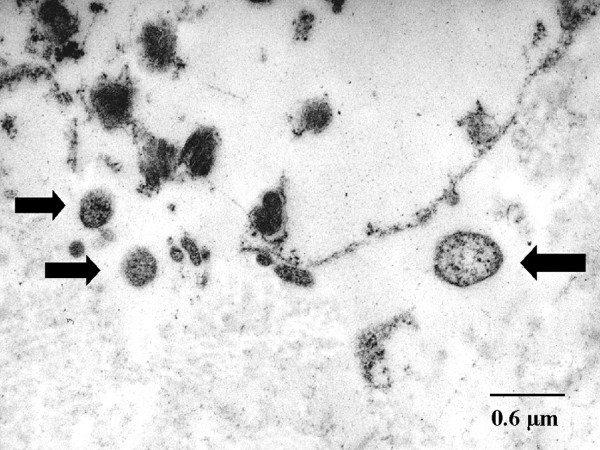
Transmission electron microscopy of alpha-1 antitrypsin deficiency emphysema, Chlamydial bodies shown by arrows, destruction of the interstitial connective tissue, Ultrastructure less well preserved after fixation of formaldehyde Contributed by Anhenn O et al. (published in BMC Infect Dis, v.4; 2004)
(Click Image to Enlarge)
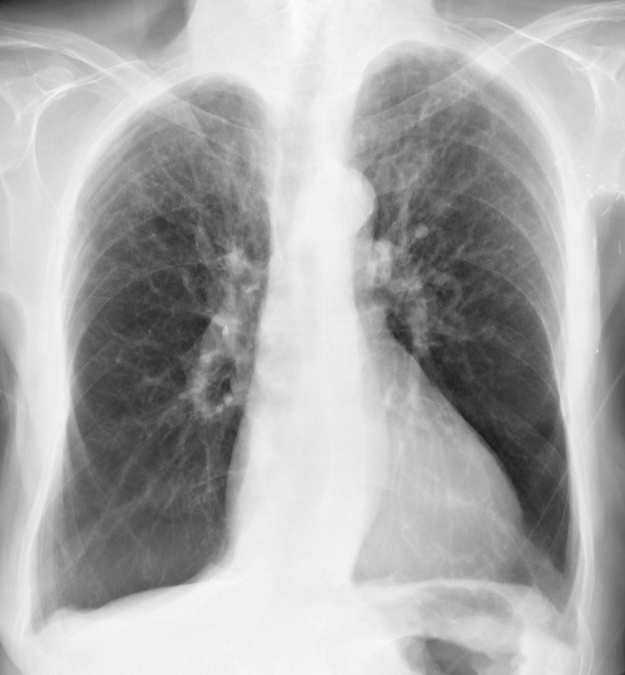
X-ray, COPD, Chronic Obstructive Pulmonary Disease, Alpha-1 Antitrypsin Deficiency Contributed by chestatlas.com (H. Shulman MD)
References
Hersh CP, Campbell EJ, Scott LR, Raby BA. Alpha-1 Antitrypsin Deficiency as an Incidental Finding in Clinical Genetic Testing. American journal of respiratory and critical care medicine. 2019 Jan 15:199(2):246-248. doi: 10.1164/rccm.201809-1679LE. Epub [PubMed PMID: 30359090]
Tasch JJ, McLaughlan AT, Nasir AA. A Novel Approach to Screening for Alpha-1 Antitrypsin Deficiency: Inpatient Testing at a Teaching Institution. Chronic obstructive pulmonary diseases (Miami, Fla.). 2018 Apr 1:5(2):106-110. doi: 10.15326/jcopdf.5.2.2017.0170. Epub 2018 Apr 1 [PubMed PMID: 30374448]
Gramegna A, Aliberti S, Confalonieri M, Corsico A, Richeldi L, Vancheri C, Blasi F. Alpha-1 antitrypsin deficiency as a common treatable mechanism in chronic respiratory disorders and for conditions different from pulmonary emphysema? A commentary on the new European Respiratory Society statement. Multidisciplinary respiratory medicine. 2018:13():39. doi: 10.1186/s40248-018-0153-4. Epub 2018 Oct 8 [PubMed PMID: 30338069]
Level 3 (low-level) evidenceAmerican Thoracic Society, European Respiratory Society. American Thoracic Society/European Respiratory Society statement: standards for the diagnosis and management of individuals with alpha-1 antitrypsin deficiency. American journal of respiratory and critical care medicine. 2003 Oct 1:168(7):818-900 [PubMed PMID: 14522813]
Owen MC, Brennan SO, Lewis JH, Carrell RW. Mutation of antitrypsin to antithrombin. alpha 1-antitrypsin Pittsburgh (358 Met leads to Arg), a fatal bleeding disorder. The New England journal of medicine. 1983 Sep 22:309(12):694-8 [PubMed PMID: 6604220]
Level 3 (low-level) evidenceOkayama H, Brantly M, Holmes M, Crystal RG. Characterization of the molecular basis of the alpha 1-antitrypsin F allele. American journal of human genetics. 1991 Jun:48(6):1154-8 [PubMed PMID: 2035534]
Miravitlles M, Turner AM, Torres-Duran M, Tanash H, Rodríguez-García C, López-Campos JL, Chlumsky J, Guimaraes C, Rodríguez-Hermosa JL, Corsico A, Martinez-González C, Hernández-Pérez JM, Bustamante A, Parr DG, Casas-Maldonado F, Hecimovic A, Janssens W, Lara B, Barrecheguren M, González C, Stolk J, Esquinas C, Clarenbach CF, EARCO study investigators. Characteristics of individuals with alpha-1 antitrypsin deficiency from Northern and Southern European countries: EARCO international registry. The European respiratory journal. 2023 Mar:61(3):. pii: 2201949. doi: 10.1183/13993003.01949-2022. Epub 2023 Mar 30 [PubMed PMID: 36997232]
Curjuric I, Imboden M, Bettschart R, Caviezel S, Dratva J, Pons M, Rothe T, Schmidt-Trucksäss A, Stolz D, Thun GA, von Eckardstein A, Kronenberg F, Ferrarotti I, Probst-Hensch NM. Alpha-1 antitrypsin deficiency: From the lung to the heart? Atherosclerosis. 2018 Mar:270():166-172. doi: 10.1016/j.atherosclerosis.2018.01.042. Epub 2018 Jan 31 [PubMed PMID: 29432934]
McCarthy C, Lara Gallego B, Trapnell BC, McCormack FX. Epidemiology of Rare Lung Diseases: The Challenges and Opportunities to Improve Research and Knowledge. Advances in experimental medicine and biology. 2017:1031():419-442. doi: 10.1007/978-3-319-67144-4_24. Epub [PubMed PMID: 29214586]
Level 3 (low-level) evidenceStrnad P, McElvaney NG, Lomas DA. Alpha(1)-Antitrypsin Deficiency. The New England journal of medicine. 2020 Apr 9:382(15):1443-1455. doi: 10.1056/NEJMra1910234. Epub [PubMed PMID: 32268028]
Stoller JK, Aboussouan LS. A review of α1-antitrypsin deficiency. American journal of respiratory and critical care medicine. 2012 Feb 1:185(3):246-59. doi: 10.1164/rccm.201108-1428CI. Epub 2011 Sep 29 [PubMed PMID: 21960536]
Mahadeva R, Chang WS, Dafforn TR, Oakley DJ, Foreman RC, Calvin J, Wight DG, Lomas DA. Heteropolymerization of S, I, and Z alpha1-antitrypsin and liver cirrhosis. The Journal of clinical investigation. 1999 Apr:103(7):999-1006 [PubMed PMID: 10194472]
Level 3 (low-level) evidenceMiravitlles M, Dirksen A, Ferrarotti I, Koblizek V, Lange P, Mahadeva R, McElvaney NG, Parr D, Piitulainen E, Roche N, Stolk J, Thabut G, Turner A, Vogelmeier C, Stockley RA. European Respiratory Society statement: diagnosis and treatment of pulmonary disease in α(1)-antitrypsin deficiency. The European respiratory journal. 2017 Nov:50(5):. pii: 1700610. doi: 10.1183/13993003.00610-2017. Epub 2017 Nov 30 [PubMed PMID: 29191952]
. A registry of patients with severe deficiency of alpha 1-antitrypsin. Design and methods. The Alpha 1-Antitrypsin Deficiency Registry Study Group. Chest. 1994 Oct:106(4):1223-32 [PubMed PMID: 7924498]
Level 2 (mid-level) evidenceSt Jean P, Hart B, Webster M, Steed D, Adamson J, Powell J, Ferrell R. Alpha-1-antitrypsin deficiency in aneurysmal disease. Human heredity. 1996 Mar-Apr:46(2):92-7 [PubMed PMID: 8666418]
Schievink WI, Puumala MR, Meyer FB, Raffel C, Katzmann JA, Parisi JE. Giant intracranial aneurysm and fibromuscular dysplasia in an adolescent with alpha 1-antitrypsin deficiency. Journal of neurosurgery. 1996 Sep:85(3):503-6 [PubMed PMID: 8751640]
Level 3 (low-level) evidenceFrederick WG, Enriquez R, Bookbinder MJ. Peripheral neuropathy associated with alpha 1-antitrypsin deficiency. Archives of neurology. 1990 Feb:47(2):233-5 [PubMed PMID: 2154173]
Level 3 (low-level) evidenceCampos MA, Wanner A, Zhang G, Sandhaus RA. Trends in the diagnosis of symptomatic patients with alpha1-antitrypsin deficiency between 1968 and 2003. Chest. 2005 Sep:128(3):1179-86 [PubMed PMID: 16162704]
Level 2 (mid-level) evidencePatel D, Teckman JH. Alpha-1-Antitrypsin Deficiency Liver Disease. Clinics in liver disease. 2018 Nov:22(4):643-655. doi: 10.1016/j.cld.2018.06.010. Epub 2018 Aug 22 [PubMed PMID: 30266154]
Torres-Durán M, Lopez-Campos JL, Barrecheguren M, Miravitlles M, Martinez-Delgado B, Castillo S, Escribano A, Baloira A, Navarro-Garcia MM, Pellicer D, Bañuls L, Magallón M, Casas F, Dasí F. Alpha-1 antitrypsin deficiency: outstanding questions and future directions. Orphanet journal of rare diseases. 2018 Jul 11:13(1):114. doi: 10.1186/s13023-018-0856-9. Epub 2018 Jul 11 [PubMed PMID: 29996870]
Level 3 (low-level) evidenceMayer AS, Stoller JK, Bucher Bartelson B, James Ruttenber A, Sandhaus RA, Newman LS. Occupational exposure risks in individuals with PI*Z alpha(1)-antitrypsin deficiency. American journal of respiratory and critical care medicine. 2000 Aug:162(2 Pt 1):553-8 [PubMed PMID: 10934086]
Level 2 (mid-level) evidenceTamondong-Lachica DR, Skolnik N, Hurst JR, Marchetti N, Rabe APJ, Montes de Oca M, Celli BR. GOLD 2023 Update: Implications for Clinical Practice. International journal of chronic obstructive pulmonary disease. 2023:18():745-754. doi: 10.2147/COPD.S404690. Epub 2023 May 5 [PubMed PMID: 37180752]
Walters JA, Tang JN, Poole P, Wood-Baker R. Pneumococcal vaccines for preventing pneumonia in chronic obstructive pulmonary disease. The Cochrane database of systematic reviews. 2017 Jan 24:1(1):CD001390. doi: 10.1002/14651858.CD001390.pub4. Epub 2017 Jan 24 [PubMed PMID: 28116747]
Level 1 (high-level) evidenceAlison JA, McKeough ZJ, Johnston K, McNamara RJ, Spencer LM, Jenkins SC, Hill CJ, McDonald VM, Frith P, Cafarella P, Brooke M, Cameron-Tucker HL, Candy S, Cecins N, Chan AS, Dale MT, Dowman LM, Granger C, Halloran S, Jung P, Lee AL, Leung R, Matulick T, Osadnik C, Roberts M, Walsh J, Wootton S, Holland AE, Lung Foundation Australia and the Thoracic Society of Australia and New Zealand. Australian and New Zealand Pulmonary Rehabilitation Guidelines. Respirology (Carlton, Vic.). 2017 May:22(4):800-819. doi: 10.1111/resp.13025. Epub 2017 Mar 24 [PubMed PMID: 28339144]
Luna Diaz LV, Iupe I, Zavala B, Balestrini KC, Guerrero A, Holt G, Calderon-Candelario R, Mirsaeidi M, Campos M. Improving adherence to alpha-1 antitrypsin deficiency screening guidelines using the pulmonary function laboratory. International journal of chronic obstructive pulmonary disease. 2017:12():2257-2259. doi: 10.2147/COPD.S143424. Epub 2017 Jul 31 [PubMed PMID: 28814853]
Campos M, Lascano J. Therapeutics: Alpha-1 Antitrypsin Augmentation Therapy. Methods in molecular biology (Clifton, N.J.). 2017:1639():249-262. doi: 10.1007/978-1-4939-7163-3_25. Epub [PubMed PMID: 28752465]
Kueppers F, Sanders C. State-of-the-art testing for alpha-1 antitrypsin deficiency. Allergy and asthma proceedings. 2017 Mar 24:38(2):108-114. doi: 10.2500/aap.2017.38.4031. Epub 2017 Jan 24 [PubMed PMID: 28120746]
McElvaney NG, Burdon J, Holmes M, Glanville A, Wark PA, Thompson PJ, Hernandez P, Chlumsky J, Teschler H, Ficker JH, Seersholm N, Altraja A, Mäkitaro R, Chorostowska-Wynimko J, Sanak M, Stoicescu PI, Piitulainen E, Vit O, Wencker M, Tortorici MA, Fries M, Edelman JM, Chapman KR, RAPID Extension Trial Group. Long-term efficacy and safety of α1 proteinase inhibitor treatment for emphysema caused by severe α1 antitrypsin deficiency: an open-label extension trial (RAPID-OLE). The Lancet. Respiratory medicine. 2017 Jan:5(1):51-60. doi: 10.1016/S2213-2600(16)30430-1. Epub 2016 Dec 2 [PubMed PMID: 27916480]
Dummer J, Dobler CC, Holmes M, Chambers D, Yang IA, Parkin L, Smith S, Wark P, Dev A, Hodge S, Dabscheck E, Gooi J, Samuel S, Knowles S, Holland AE. Diagnosis and treatment of lung disease associated with alpha one-antitrypsin deficiency: A position statement from the Thoracic Society of Australia and New Zealand. Respirology (Carlton, Vic.). 2020 Mar:25(3):321-335. doi: 10.1111/resp.13774. Epub 2020 Feb 6 [PubMed PMID: 32030868]
Nelson DR, Teckman J, Di Bisceglie AM, Brenner DA. Diagnosis and management of patients with α1-antitrypsin (A1AT) deficiency. Clinical gastroenterology and hepatology : the official clinical practice journal of the American Gastroenterological Association. 2012 Jun:10(6):575-80. doi: 10.1016/j.cgh.2011.12.028. Epub 2011 Dec 23 [PubMed PMID: 22200689]
Wilke A, Semper H, Gross C, Grohé C. [Longterm Homecare Augmentation Program in Alpha-1-Antitrypsin Deficient Patients]. Pneumologie (Stuttgart, Germany). 2018 Aug:72(8):590-597. doi: 10.1055/a-0618-7493. Epub 2018 Aug 8 [PubMed PMID: 30089330]
Stockley RA, Edgar RG, Starkey S, Turner AM. Health status decline in α-1 antitrypsin deficiency: a feasible outcome for disease modifying therapies? Respiratory research. 2018 Jul 20:19(1):137. doi: 10.1186/s12931-018-0844-6. Epub 2018 Jul 20 [PubMed PMID: 30029692]
Silverman EK, Sandhaus RA. Clinical practice. Alpha1-antitrypsin deficiency. The New England journal of medicine. 2009 Jun 25:360(26):2749-57. doi: 10.1056/NEJMcp0900449. Epub [PubMed PMID: 19553648]