Introduction
Tangier disease is an inherited condition, first described in two siblings from the Tangier island of Virginia in the Chesapeake Bay in 1961 and named after the island. There is a significant decrease in HDL, which is also known as the good cholesterol, and accumulation of cholesterol esters in many organs within the body, particularly the reticuloendothelial systems. These organs will appear yellow-orange in color and larger in size. That is why these patients will have an increased risk of premature cardiovascular events at a younger age than normal individuals.[1]
Clinically, it is characterized by enlargement of the liver, spleen, lymph nodes, and tonsils associated with peripheral neuropathy in children and adolescents.
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
Tangier disease exhibits an autosomal recessive pattern for its clinical features while exhibits an autosomal codominant trait for its biochemical findings. Therefore, if the patient received both the recessive genes from their parents, they will have an extremely low level of HDL, and If the patient inherited only one gene from their parents, they would have a relatively low level of HDL (about half the normal level in normal individuals) but no overt clinical manifestation of the disease. Heterogeneous parents for the pathology will have a 25% chance of having an affected sibling or homozygous sibling, 50% of having a carrier sibling or heterozygous sibling, and 25% of having a normal sibling, while homozygous parent married to a normal individual will have 100% carrier siblings.
Mutations have been found in chromosome 9q31, which encodes for the ATP-Binding cassette transporter (ABCA-1) gene. These transporters normally transmit cholesterol and phospholipids from inside the cells into the liver. Thus cholesterol will be accumulated within the cells, which eventually impairs cell functions and lead to their death.[2][3]
Epidemiology
The prevalence of the disease has been identified as 1/1,000,000. The disease is rare, and only about 100 cases have been identified since 1961. It has been described in persons of all races. Conseguanity marriages show an increase in the risk of developing the disease in the following generation. However, two reported cases of the disease are described in children from families unrelated to each other.[4]
Pathophysiology
The HDL life cycle starts inside the cells when very small discoidal Pre beta-1 HDL picks up free cholesterol from cells with the help of ABCA transporter, defect in this stage will result in "Tangier disease." This discoid shaped nascent HDL then undergoes esterification via the enzyme lecithin cholesterol acyltransferase (LCAT) converting it to a mature HDL, defect in this stage will result in "familial LCAT deficiency." These mature HDL particles with the help of cholesterol ester transfer protein(CEPT) will mediate the transfer of cholesterol esters to triglyceride-rich lipoprotein particles (VLDL, IDL, LDL) in exchange for triglyceride. This remaining cholesterol-rich HDL will be preferred for the use inside the liver where it is either recycled or transferred to the HDL cycle again through destroying into small discoidal particles again or is catabolized by the liver.[4]
Histopathology
Splenomegaly is accompanied by mild thrombocytopenia (with a platelet count of 30,000 to 120,000 cells per microliter) and reticulocytosis in many patients. The appearance of the rectal mucosa is usually abnormal and maybe the most reliable physical finding when the palatine and pharyngeal tonsils have been completely removed. Ocular abnormalities in Tangier disease include corneal opacity, ectropion, the mottled retinal pigment in the macula, and/or periphery.
History and Physical
Patients who inherited a heterozygous gene for the disease will have relatively low HDL with no deposition of cholesterol esters within body organs. In contrast, individuals who inherited both the recessive genes will have a cholesterol deposition manifestation and an extremely low HDL.
Clinical Findings:
- Oral cavity: Cgolesterol deposition in tonsils is one the salient features. Cholesterol deposits appear yellow/orange in color, significantly enlarged in children and young adults—usually the first observed presentation.
- Cardiovascular system: Affected people will have premature atherosclerosis; therefore, they are more prone to coronary artery diseases, and strokes usually occur in an early stage of their age. Sometimes heart valves could also be affected.[5]
- Nervous system: deposition of cholesterols in peripheral nerves inside Schwann cells cause demyelination of nerve fibers, which can appear in different manifestations: Syringomyelia-like neuropathy (loss of pain and temperature sensation mainly affecting the upper extremities), multifocal sensorial and motor neuropathy(relapsing-remitting pattern) and distal symmetric polyneuropathy subtype.[6]
- Cornea: The clouding of the cornea has been reported and usually mild with no vision loss.
- Reticuloendothelial System: cholesterol deposition in these organs leads to hepatosplenomegaly. The involvement of spleen can also lead to thrombocytopenia. It also could be deposited within the lymph nodes.
- Pancreas: Diabetes has been reported due to deposition inside the Alpha cells of the pancreas.
- Hematologic manifestations: They include thrombocytopenia, reticulocytosis, hemolytic anemia, and stomatocytosis.
Tangier disease appears earlier in life in the 1st decade as a tonsillar enlargement and clouding of the cornea. Neuropathy and cardiovascular disease are the most devastating developments caused by Tangier's disease.[7]
There are other clinical signs that can be found:
- Abdominal pain
- Chronic noninfectious lymphadenopathy
- Dry skin
- Nail dystrophy
- Facial diplegia
Evaluation
The diagnosis of Tangier disease requires a high index of suspicion. Besides clinical findings which could appear earlier in the first decades or wait till the second or third decades. Blood values remain the mainstay diagnostic tools to make a diagnosis. The patient regularly presented with Low HDL (less than 5 mg/dl ), a low Apo-a1 level below 5, besides Low total plasma cholesterol (less than 150mg/dl). Another blood findings include a normal or elevated Triglyceride level, decreased LDL level as there will be upregulation in LDL receptor expression and thrombocytopenia.[8]
The gold standard for the diagnosis of Tangier disease is ABCA1 molecular gene sequencing. If genetic testing cannot be offered, then tissue biopsy could also be taken on multiple organs of the body: bone marrow, jejunum, liver, and the rectum, which is considered the site of choice for biopsy and they would show huge collections of cholesterol-laden macrophages.
Measurements could be done to assess multiple organ systems involve: Nerve conduction studies and EMG to determine the presence or extent of peripheral neuropathy, ophthalmologic evaluation to assess for any corneal opacities, abdominal ultrasound to assess for hepatosplenomegaly, carotid duplex for any carotid plaques and CT angiography to assess for coronary atherosclerosis.
High-risk individuals include relatives of known diagnostic cases, should be assessed for the presence of a carrier state of the mutated gene. Also, the prenatal diagnosis could be offered. It is also appropriate to follow up people at risk through checking their lipid profile (total cholesterol, HDL-cholesterol, triglyceride, and calculated LDL-cholesterol) and apo A-I concentration. Heterozygotes carriers usually have no clinical symptoms and are characterized at the biochemical level by half-normal serum levels of HDL.[3]
Antenatal diagnosis is possible, although it is not usually done.
Treatment / Management
There is no exact treatment in the meantime for Tangier disease, and the attempts are to increase HDL through lifestyle modifications through maintaining regular aerobic exercise, maintaining a healthy weight, smoking cessation, and replacement of monounsaturated for saturated fatty acids raise HDL cholesterol. These interventions could improve the symptoms, especially the peripheral neuropathies. There is no firm evidence of the benefit from drug therapy to target low HDL cholesterol and optimize their LDL level, but lipid-lowering agents such as statins, niacin, and fibrates can be given either alone or in combinations.[8]
The treatment is administered based on the manifestation such as tonsillectomy in case of tonsil enlargement leading to mass symptoms or airway obstruction. Corneal transplantation may be required in case of corneal opacities, transient bracing (such as ankle-foot orthosis), and exercise for those with peripheral neuropathy. Precautions such as avoidance of high-impact sports or activities should be thoroughly prescribed in order to prevent splenic rupture if the patient has splenomegaly.[4](B3)
A future therapeutic approach is suggested through upregulating cholesterol uptake by hepatocytes or downregulating HDL metabolism. This can be achieved by targeted gene therapy, which causes ABC1 overexpression leading to increase cholesterol pick up within the cells.
Differential Diagnosis
Many inherited diseases that present with manifestations of low HDL level and peripheral neuropathy should be put into consideration:
- Familial HDL deficiency is an autosomal dominant disorder associated with very low serum HDL concentrations and premature CHD but none of the systemic findings that present in Tangier disease exist in it.[9]
- Abetalipoproteinemia is a rare autosomal recessive disorder that is characterized by defective assembly and secretion of apolipoprotein B (Apo B) and apo B containing lipoproteins. A spectrum of clinical manifestations can be seen include failure to thrive and fat malabsorptions. Later clinical outcomes include retinitis pigmentosa, peripheral neuropathy, and ataxia.[10]
- Niemann-Pick disease is an autosomal recessive lysosome storage disease that occurs due to the accumulation of abnormal metabolic products. Usually has far greater hepatosplenomegaly than in Tangier disease, progressive neurodegeneration, and cherry-red spots on the macula.[11]
- LCAT deficiency is an autosomal recessive due to Lecithin cholesterol acyltransferase deficiency. presents with annular corneal opacity, and progressive renal disease with proteinuria.[12]
- Fish-Eye disease is an autosomal recessive due to a partial deficiency of LCAT. It manifests with corneal opacity with normal renal function.[13]
- Charcot-Marie-Tooth Disease is an autosomal recessive disease presented with peripheral neuropathy and different types of foot deformities.[14]
- Other causes that make a low HDL including taking Drugs such as beta-blockers, benzodiazepines, and anabolic steroids. Also,Acute infection(HIV) , inflammation, or gammopathies( multiple myeloma).
Prognosis
Tangier disease is a rare disease, and there are insufficient data to determine the life expectancy of these patients. However, Prognosis is usually good and depends mainly on the progression of atherosclerosis and peripheral neuropathies, and patients inherited the homogenous genes for Tangier disease will have HDL cholesterol of less than 5 mg/dl are more prone to coronary artery disease and should have a regular follow up with their doctors.[15]
Complications
Despite the substantial evidence of an inverse relationship between cardiovascular event risk and high-density lipoprotein (HDL) cholesterol, low levels of HDL cholesterol have not been established as causative of this relationship or with the development of atherosclerosis. However, the deposition of cholesterol esters merits consideration as the major risk factor through the development of premature cardiovascular events in Tangier disease patients, but these mostly occur after the age of 40. Patients having hepatosplenomegaly may be presented with splenic rupture in case of practicing aggressive contact sports. Diabetes mellitus could result from cholesterol deposition in the pancreas. Airway obstruction or mass symptoms in massive tonsillar enlargement. Peripheral neuropathies could be aggressive and in relapsing-remitting stages and could affect any part of the nervous system. At last, mild to moderate visual impairment is also reported.[15]
Deterrence and Patient Education
HDL or good cholesterol plays an important role in preventing cardiovascular events through its main function of transmitting cholesterol from the peripheral tissues to be excreted by the liver. It has many other functions:
- It plays as an antioxidant and anti-inflammatory agent through the inhibition of LDL oxidation and endothelial adhesion molecules expression.
- It has antithrombotic properties.
- It can also improve endothelial function through the promotion of cells repair and promote angiogenesis.
- It helps diabetic patients to have better diabetic control.[16]
To maintain a healthy lifestyle and to keep lipids profile within the regular limit there are many advises to be given include: regular aerobic exercise, maintaining a healthy weight, smoking cessation, and replacement of monounsaturated for saturated fatty acids raise HDL cholesterol. There are also certain types of food which are recommended for patients that are at risk for a CVS events that are rich in omega-3 fatty acids and could increase the HDL level and lower the LDL level includes; Olive oil, whole grains, beans and legumes, High fiber fruits, fatty fish, Flax and chia seeds, nuts and avocado.[17][18]
Some medications, such as beta-blockers, benzodiazepines, and androgens, are associated with a decrease in HDL cholesterol. We do not stop these drugs if they are important to the treatment of other medical conditions. Also, medications that are toxic or known to cause peripheral neuropathy, such as vincristine or taxols (paclitaxel), should be used with caution.
Pearls and Other Issues
In patients undergoing thoracic cardiac surgery for coronary stenosis and angina pectoris, there is a likelihood that the patient may undergo other interventions (when possible) due to recurrent stenosis.[19][20]
Enhancing Healthcare Team Outcomes
Managing a patient with Tangier disease requires a close watch to the patient for any progression in his symptoms to make the right move in a perfect timely manner. A couple of specialty doctors should interact to take care of this patient by frequent visits to an ophthalmologist to assess for any visual impairment, a cardiology assessment started at adulthood to evaluate for any atherosclerotic changes, a neurologist to monitor any neurological manifestations, and hematologist to evaluate anemic and thrombocytopenic states.[8]
Genetic Counseling should be offered to individuals and families of affected patients describing the nature of the disease, the mode of inheritance, manifestations, and its complications. And prenatal genetic testing should be offered.
Because of the rarity of the disorder, there is a lack of randomized clinical trials on the long-term benefits of many drugs and the disease itself. However, studies indicate that when patients are diagnosed and referred early that specializes in the management of this lipid disorder, the outcomes are improved.
Media
(Click Image to Enlarge)
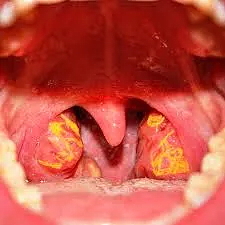
In Tangier's disease, the tonsils (as in the photo) appear enlarged and with a typical yellow-orange color. Contributed by Bruno Bordoni, PhD.
References
Burnett JR,Hooper AJ,McCormick SPA,Hegele RA, Tangier Disease 1993; [PubMed PMID: 31751110]
Ducasa GM,Mitrofanova A,Mallela SK,Liu X,Molina J,Sloan A,Pedigo CE,Ge M,Santos JV,Hernandez Y,Kim JJ,Maugeais C,Mendez AJ,Nair V,Kretzler M,Burke GW,Nelson RG,Ishimoto Y,Inagi R,Banerjee S,Liu S,Szeto HH,Merscher S,Fontanesi F,Fornoni A, ATP-binding cassette A1 deficiency causes cardiolipin-driven mitochondrial dysfunction in podocytes. The Journal of clinical investigation. 2019 Jul 22; [PubMed PMID: 31329164]
Schaefer EJ,Kay LL,Zech LA,Brewer HB Jr, Tangier disease. High density lipoprotein deficiency due to defective metabolism of an abnormal apolipoprotein A-i (ApoA-ITangier). The Journal of clinical investigation. 1982 Nov; [PubMed PMID: 7130397]
Puntoni M,Sbrana F,Bigazzi F,Sampietro T, Tangier disease: epidemiology, pathophysiology, and management. American journal of cardiovascular drugs : drugs, devices, and other interventions. 2012 Oct 1; [PubMed PMID: 22913675]
Level 3 (low-level) evidenceSerfaty-Lacrosniere C,Civeira F,Lanzberg A,Isaia P,Berg J,Janus ED,Smith MP Jr,Pritchard PH,Frohlich J,Lees RS, Homozygous Tangier disease and cardiovascular disease. Atherosclerosis. 1994 May; [PubMed PMID: 7945562]
Level 3 (low-level) evidenceMercan M,Yayla V,Altinay S,Seyhan S, Peripheral neuropathy in Tangier disease: A literature review and assessment. Journal of the peripheral nervous system : JPNS. 2018 Jun; [PubMed PMID: 29582519]
Kolovou GD,Mikhailidis DP,Anagnostopoulou KK,Daskalopoulou SS,Cokkinos DV, Tangier disease four decades of research: a reflection of the importance of HDL. Current medicinal chemistry. 2006; [PubMed PMID: 16611066]
Pervaiz MA,Gau G,Jaffe AS,Saenger AK,Baudhuin L,Ellison J, A Non-classical Presentation of Tangier Disease with Three ABCA1 Mutations. JIMD reports. 2012; [PubMed PMID: 23430904]
Pisciotta L,Vitali C,Favari E,Fossa P,Adorni MP,Leone D,Artom N,Fresa R,Calabresi L,Calandra S,Bertolini S, A complex phenotype in a child with familial HDL deficiency due to a novel frameshift mutation in APOA1 gene (apoA-IGuastalla). Journal of clinical lipidology. 2015 Nov-Dec; [PubMed PMID: 26687706]
Junaid Z,Patel K, Abetalipoproteinemia 2020 Jan; [PubMed PMID: 30020727]
Bajwa H,Azhar W, Niemann-Pick Disease 2020 Jan; [PubMed PMID: 32310589]
Althaf MM,Almana H,Abdelfadiel A,Amer SM,Al-Hussain TO, Familial lecithin-cholesterol acyltransferase (LCAT) deficiency; a differential of proteinuria. Journal of nephropathology. 2015 Jan; [PubMed PMID: 25657982]
Level 3 (low-level) evidenceKuivenhoven JA,van Voorst tot Voorst EJ,Wiebusch H,Marcovina SM,Funke H,Assmann G,Pritchard PH,Kastelein JJ, A unique genetic and biochemical presentation of fish-eye disease. The Journal of clinical investigation. 1995 Dec; [PubMed PMID: 8675648]
Level 3 (low-level) evidenceAdam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, Bird TD. Charcot-Marie-Tooth Hereditary Neuropathy Overview. GeneReviews(®). 1993:(): [PubMed PMID: 20301532]
Level 3 (low-level) evidenceLiang Z,Li W,Yang S,Liu Z,Sun X,Gao X,Yu G, Tangier disease may cause early onset of atherosclerotic cerebral infarction: A case report. Medicine. 2018 Sep; [PubMed PMID: 30278532]
Level 3 (low-level) evidencevon Eckardstein A,Huang Y,Assmann G, Physiological role and clinical relevance of high-density lipoprotein subclasses. Current opinion in lipidology. 1994 Dec; [PubMed PMID: 7712045]
Level 3 (low-level) evidenceRondanelli M,Giacosa A,Morazzoni P,Guido D,Grassi M,Morandi G,Bologna C,Riva A,Allegrini P,Perna S, MediterrAsian Diet Products That Could Raise HDL-Cholesterol: A Systematic Review. BioMed research international. 2016; [PubMed PMID: 27882320]
Level 1 (high-level) evidenceYanai H,Tada N, Which Nutritional Factors Are Good for HDL? Journal of clinical medicine research. 2018 Dec; [PubMed PMID: 30425767]
Takami H,Kobayashi T,Nakagawa T,Sakurai M,Awata N,Yamashita S, Coronary artery bypass grafting for a patient with Tangier disease. The Japanese journal of thoracic and cardiovascular surgery : official publication of the Japanese Association for Thoracic Surgery = Nihon Kyobu Geka Gakkai zasshi. 2002 Sep [PubMed PMID: 12382407]
Level 3 (low-level) evidenceMuratsu J,Koseki M,Masuda D,Yasuga Y,Tomoyama S,Ataka K,Yagi Y,Nakagawa A,Hamada H,Fujita S,Hattori H,Ohama T,Nishida M,Hiraoka H,Matsuzawa Y,Yamashita S, Accelerated Atherogenicity in Tangier Disease. Journal of atherosclerosis and thrombosis. 2018 Oct 1 [PubMed PMID: 29563393]