Introduction
Coarctation of the aorta is a narrowing of the aorta, most commonly occurring just beyond the left subclavian artery. However, it can occur in various other locations of the aortic arch (proximal transverse) or even in the thoracic or abdominal aorta. The narrowing of the aorta raises the upper body blood pressure, causing upper extremity hypertension. Unrepaired coarctation leads to premature coronary artery disease, ventricular dysfunction, aortic aneurysm/dissection, and cerebral vascular disease by the third or fourth decade of life.[1][2][3]
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
The most common etiology of coarctation of the aorta is constriction of the aorta in the region of the patent ductus arteriosus or ductal ligamentum. The ductal tissue is thought to cause constriction in the adjacent region of the aorta. This narrows the lumen of the aorta. Coarctation can also be more complex and present as aortic arch hypoplasia and as a component of other left-sided heart lesions (mitral stenosis, aortic stenosis, hypoplastic left heart syndrome). Mid-thoracic coarctation can occur with mid-aortic syndromes. Over time, the body compensates by developing collaterals around the coarctation segment.[4]
Epidemiology
Coarctation is found in 6% to 8% of patients with congenital heart disease or approximately 0.06% to 0.08% of the general population. Coarctation of the aorta accounts for a percentage of children diagnosed with systemic hypertension. Patients with Turner syndrome (XO) have an increased risk of left-sided obstructed heart disease that includes isolated coarctation, and karyotype screening is recommended for females with the diagnosis of coarctation. The bicuspid aortic valve is commonly associated with coarctation of the aorta. Offspring and other first-degree relatives diagnosed with an obstructive left-sided cardiac lesion are at ten times the risk of coarctation and other cardiac lesions.[5]
Pathophysiology
Coarctation of the aorta causes an increase in the upper extremity blood pressure, resulting in two common presentations. The first is the neonatal presentation associated with left ventricular dysfunction and shock from the neonatal myocardium's intolerance of the sudden increase in afterload that occurs with the closure of the ductus arteriosus. This presentation often occurs within the first one to two weeks after birth. In patients with neonatal coarctation evolving while the patent ductus arteriosus is closing, the lower extremity saturation can be low as perfusion to the lower body can be maintained by ductal patency. In the era of lower extremity pulse oximetry screening in newborns, a neonate could often pass with an acceptable saturation as it is less common for the ductus to contribute significantly unless other left heart structures are hypoplastic. The second presentation occurs in older children and adults. Coarctation of the aorta in this scenario results in upper extremity hypertension, leading to early coronary artery disease, aortic aneurysm, and cerebrovascular disease.
History and Physical
The most important physical finding is upper extremity hypertension. In neonates, there can be a history of poor feeding, evidence of shock with poor perfusion, gallop heard on auscultation, and a murmur of mitral regurgitation. In significant coarctation, the femoral and dorsal pedis artery pulsations are difficult to palpate, and there is associated brachiofemoral delay. Upper extremity hypertension is most often present, and four extremity blood pressure measurements should be performed in all children and young adults being evaluated for this condition. In older children, a systolic murmur with diastolic continuation can be heard in the left infrascapular region. This murmur can represent blood flow across the coarctation or through dilated collaterals. A systolic ejection click and systolic ejection murmur in the left upper sternal border is a consistent exam finding with a bicuspid aortic valve. It is very rare in today's era for an adult to present with aortic dissection or cerebrovascular accident from an undiagnosed coarctation of the aorta.
Evaluation
The EKG in a patient with coarctation can demonstrate increased voltage in the lateral precordial leads consistent with left ventricular hypertrophy. The echocardiogram will demonstrate left ventricular hypertrophy. In neonates, the left ventricular function can be diminished. There also can be mitral regurgitation and left atrial dilation from elevated left atrial pressures. The echocardiogram can demonstrate a narrowing in the aortic arch at the level of the isthmus (just beyond the left subclavian) with Doppler velocities increased in this region, but the arch can also appear hypoplastic. The abdominal aortic pulsations are decreased with diastolic runoff. CT scanning and MRI are useful to provide detailed anatomy of the aortic arch before and after treatment.[6][7]
Treatment / Management
In a neonate presenting with shock, stabilization should occur first with cardiorespiratory support. Prostaglandin E1 infusion occasionally can open the ductus arteriosus but also seems to relax the tissue of the coarctation segment. Often medical support alone will allow some recovery of ventricular function. The treatment for coarctation of the aorta is to eliminate the narrowed segment. This can be accomplished surgically or via transcatheter techniques. Surgery requires the removal of the coarctation segment and direct anastomosis of the normal aorta. The transcatheter technique utilizes balloon and stent angioplasty. Most institutions perform surgery for neonates and small children. Many institutions will perform cardiac catheterization and primary stent angioplasty in adolescents and adults. Balloon angioplasty has been performed in neonates and children. No surgical or interventional technique is a cure for coarctation. In neonates, the risk of recoarctation following surgical intervention is approximately 10%. When recoarctation occurs, balloon angioplasty is advised. There is a lifelong risk of developing an aortic aneurysm, which seems to increase when the treatment is balloon angioplasty alone. Furthermore, even after an intervention, there is an increased risk of developing essential hypertension. There also is an increased risk of cerebral aneurysms in patients with coarctation of the aorta (treated or untreated). For these reasons, patients diagnosed with coarctation of the aorta should seek lifelong follow-up with a congenital heart disease specialist.[8][3][9](A1)
Differential Diagnosis
- Aortic dissection
- Coarctation of the aorta
- Myocarditis
- Pediatric hypoplastic left heart syndrome
- Pediatric sepsis
- Peripheral arterial occlusive disease
Prognosis
- Aortic coarctation of the aorta is a lifelong disease, and the long-term prognosis is guarded. Follow-up care is vital as recurrence of coarctation and hypertension are not uncommon.
- These individuals also need prophylaxis for endocarditis if they undergo any invasive procedure.
- Short-term who fail to manage hypertension tend to have a worse outcome compared to normotensive individuals.
- Long-term data are lacking as most patients are lost to follow-up. However, survival is decreased compared to the normal population.
- Individuals who fail to get the coarctation repaired are usually dead by age 50.
Complications
- Recurrent coarctation
-
Aortic aneurysm
-
Hypertension
-
Cerebral aneurysm
-
Cardiomyopathy
-
Paralysis - the risk of postoperative paraplegia can be reduced with mild hypothermia (34 °C to 35 °C) with limited cross-clamp time
-
Post coarctectomy syndrome
Postoperative and Rehabilitation Care
Once the coarctation is repaired, the patient's blood pressure must be monitored and treated if elevated.
Deterrence and Patient Education
- Coarctation cannot be prevented, but a timely referral to a cardiologist for an echocardiogram is recommended.
- The patient should exercise regularly, not smoke, and maintain a healthy weight.
Pearls and Other Issues
Follow-up for coarctation of the aorta should be lifelong with a congenital heart specialist. Even after effective treatment, the risk of developing hypertension increases. Regardless of the chosen therapy, there is a risk of developing aortic aneurysms, and periodic aorta imaging is required. Aneurysm development is highest after balloon angioplasty alone but is also associated with surgical repair and stent implantation. Cerebral imaging could be recommended to screen for aneurysms in adults.
Enhancing Healthcare Team Outcomes
There are many recommendations for the diagnosis and management of aortic coarctation, especially in adult males and females. There is no cure for the disorder, and even after treatment, there is a risk of recurrence and development of hypertension. Thus, patients need lifelong follow-up with an interprofessional group of health professionals, including a cardiologist, cardiac surgeon, internist, and critical care consultant. After surgery, the patient must be educated by the cardiovascular nurse and pharmacist on the importance of compliance with blood pressure medications. Regular follow-up is vital to ensure that the blood pressure is controlled and the distal pulses are not diminished. The primary care provider must ensure the patient receives endocarditis prophylaxis when undergoing any invasive procedure.[10][11] [Level 3]
Outcomes
The short-term outlook for surgical patients with coarctation of the aorta is excellent, but even these patients have a guarded prognosis. Long-term data are not yet available primarily because patients are lost to follow up. Data indicate that in the long-term, recurrence of aortic coarctation, hypertension, stroke, paraplegia, and adverse cardiac events are not rare. In view of these negative observations, an interprofessional team approach is highly recommended to prevent high morbidity and mortality. [12][13][14] [Level 5]
Media
(Click Image to Enlarge)
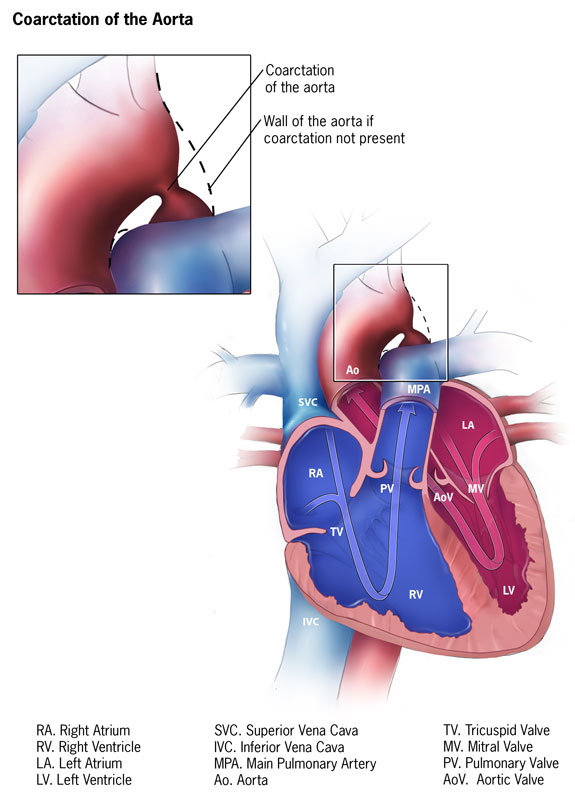
Coarctation of the Aorta Contributed by The Center for Disease Prevention and Control
(Click Image to Enlarge)
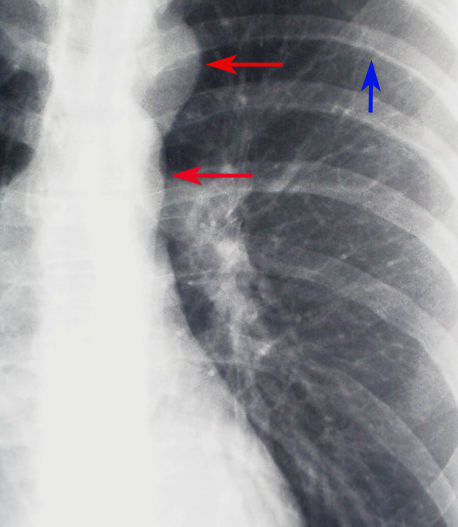
X-ray, Cardiac, Coarctation of the Aorta, Close up Contributed by chestatlas.com (H. Shulman MD)
(Click Image to Enlarge)
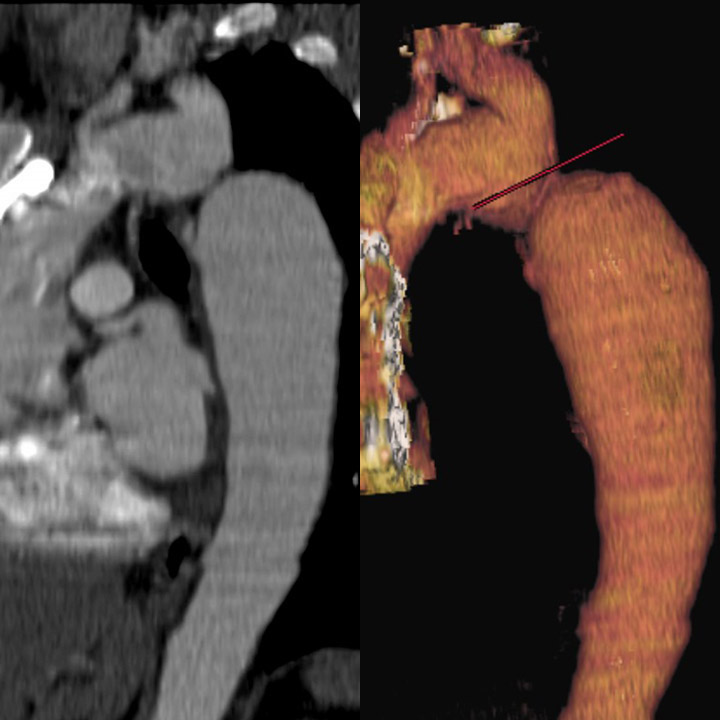
CT Scan, Magnetic Resonance, Cardiac, Coarctation of the Aorta Contributed by chestatlas.com (H. Shulman MD)
(Click Image to Enlarge)
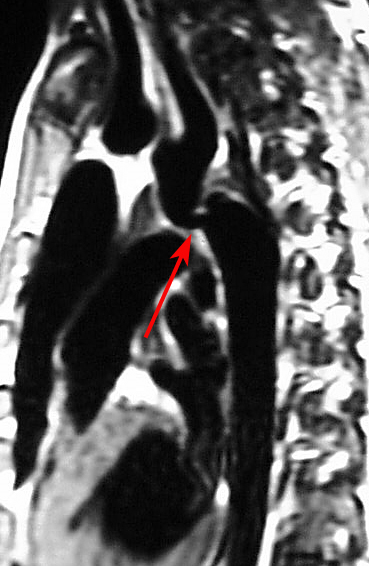
MRI, Cardiac, Coarctation of the Aorta Contributed by chestatlas.com (H. Shulman MD)
References
Hoffman JI. The challenge in diagnosing coarctation of the aorta. Cardiovascular journal of Africa. 2018 Jul/Aug 23:29(4):252-255. doi: 10.5830/CVJA-2017-053. Epub 2017 Dec 11 [PubMed PMID: 29293259]
Boris JR. Primary-care management of patients with coarctation of the aorta. Cardiology in the young. 2016 Dec:26(8):1537-1542. doi: 10.1017/S1047951116001748. Epub [PubMed PMID: 28148323]
Beckmann E, Jassar AS. Coarctation repair-redo challenges in the adults: what to do? Journal of visualized surgery. 2018:4():76. doi: 10.21037/jovs.2018.04.07. Epub 2018 Apr 23 [PubMed PMID: 29780722]
Hanneman K, Newman B, Chan F. Congenital Variants and Anomalies of the Aortic Arch. Radiographics : a review publication of the Radiological Society of North America, Inc. 2017 Jan-Feb:37(1):32-51. doi: 10.1148/rg.2017160033. Epub 2016 Nov 18 [PubMed PMID: 27860551]
Yetman AT, Starr L, Sanmann J, Wilde M, Murray M, Cramer JW. Clinical and Echocardiographic Prevalence and Detection of Congenital and Acquired Cardiac Abnormalities in Girls and Women with the Turner Syndrome. The American journal of cardiology. 2018 Jul 15:122(2):327-330. doi: 10.1016/j.amjcard.2018.03.357. Epub 2018 Apr 17 [PubMed PMID: 29731120]
Kaya U, Colak A, Becit N, Ceviz M, Kocak H. Surgical Management of Aortic Coarctation from Infant to Adult. The Eurasian journal of medicine. 2018 Feb:50(1):14-18. doi: 10.5152/eurasianjmed.2017.17273. Epub 2017 Dec 29 [PubMed PMID: 29531485]
Conti L, Borg Savona S, Spiteri T, Degiovanni J, Borg A, Caruana M. Aortic coarctation - never too late to diagnose, never too late to treat. Images in paediatric cardiology. 2017 Jul-Sep:19(3):1-11 [PubMed PMID: 29731785]
Wu Y, Jin X, Kuang H, Lv T, Li Y, Zhou Y, Wu C. Is balloon angioplasty superior to surgery in the treatment of paediatric native coarctation of the aorta: a systematic review and meta-analysis. Interactive cardiovascular and thoracic surgery. 2019 Feb 1:28(2):291-300. doi: 10.1093/icvts/ivy224. Epub [PubMed PMID: 30060099]
Level 1 (high-level) evidenceBrzezinska-Rajszys G. Stents in treatment of aortic coarctation and recoarctation in small children. International journal of cardiology. 2018 Jul 15:263():40-41. doi: 10.1016/j.ijcard.2018.03.141. Epub [PubMed PMID: 29754920]
Mühler EG, Franke A, Lepper W, Grabitz RG, Herrmann G, Klues HG, Messmer BJ, Hanrath P, von Bernuth G. [The management of adolescents and adults with congenital heart defects: 3 years experiences with interdisciplinary consultation]. Zeitschrift fur Kardiologie. 1995 Jul:84(7):532-41 [PubMed PMID: 7676723]
Rajbanshi BG, Joshi D, Pradhan S, Gautam NC, Timala R, Shakya U, Sharma A, Biswakarma G, Sharma J. Primary surgical repair of coarctation of the aorta in adolescents and adults: intermediate results and consequences of hypertension. European journal of cardio-thoracic surgery : official journal of the European Association for Cardio-thoracic Surgery. 2019 Feb 1:55(2):323-330. doi: 10.1093/ejcts/ezy228. Epub [PubMed PMID: 29933438]
Gurvitz M, Burns KM, Brindis R, Broberg CS, Daniels CJ, Fuller SM, Honein MA, Khairy P, Kuehl KS, Landzberg MJ, Mahle WT, Mann DL, Marelli A, Newburger JW, Pearson GD, Starling RC, Tringali GR, Valente AM, Wu JC, Califf RM. Emerging Research Directions in Adult Congenital Heart Disease: A Report From an NHLBI/ACHA Working Group. Journal of the American College of Cardiology. 2016 Apr 26:67(16):1956-64. doi: 10.1016/j.jacc.2016.01.062. Epub [PubMed PMID: 27102511]
Ramnarine I. Role of surgery in the management of the adult patient with coarctation of the aorta. Postgraduate medical journal. 2005 Apr:81(954):243-7 [PubMed PMID: 15811888]
Lala S, Scali ST, Feezor RJ, Chandrekashar S, Giles KA, Fatima J, Berceli SA, Back MR, Huber TS, Beaver TM, Beck AW. Outcomes of thoracic endovascular aortic repair in adult coarctation patients. Journal of vascular surgery. 2018 Feb:67(2):369-381.e2. doi: 10.1016/j.jvs.2017.06.103. Epub 2017 Sep 22 [PubMed PMID: 28947226]
Salciccioli KB, Zachariah JP. Coarctation of the Aorta: Modern Paradigms Across the Lifespan. Hypertension (Dallas, Tex. : 1979). 2023 Oct:80(10):1970-1979. doi: 10.1161/HYPERTENSIONAHA.123.19454. Epub 2023 Jul 21 [PubMed PMID: 37476999]
Pugnaloni F, Doni D, Lucente M, Fiocchi S, Capolupo I. Ductus Arteriosus in Fetal and Perinatal Life. Journal of cardiovascular development and disease. 2024 Apr 1:11(4):. doi: 10.3390/jcdd11040113. Epub 2024 Apr 1 [PubMed PMID: 38667731]
Seri A, Baral N, Yousaf A, Sriramoju A, Chinta SR, Agasthi P. Outcomes of Heart Failure Hospitalizations in Adult Patients With Coarctation of Aorta: Report From National Inpatient Sample. Current problems in cardiology. 2023 Oct:48(10):101888. doi: 10.1016/j.cpcardiol.2023.101888. Epub 2023 Jun 20 [PubMed PMID: 37343776]
Ahmadi A, Mansourian M, Sabri MR, Ghaderian M, Karimi R, Roustazadeh R. Follow-up outcomes and effectiveness of stent implantation for aortic coarctation: A systematic review and meta-analysis. Current problems in cardiology. 2024 Jun:49(6):102513. doi: 10.1016/j.cpcardiol.2024.102513. Epub 2024 Mar 29 [PubMed PMID: 38556144]
Level 1 (high-level) evidence