Introduction
Aplastic anemia refers to the syndrome of chronic primary hematopoietic failure from injury leading to diminished or absent hematopoietic precursors in the bone marrow and attendant pancytopenia.[1][2]
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
Injury to bone marrow occurs in a multitude of settings (table 1). The most common etiology, idiopathic, accounts for 65%. Fanconi anemia is the most common hereditary cause. It presents in the late first decade with pancytopenia, organ hypoplasia, and bone defects including abnormal radii, absent thumbs, and short stature. Seronegative hepatitis is responsible for 5% to 10% of total cases. Telomerase defects are found in 5% to 10% of adult-onset aplastic anemia. Some of these associations are very rare for example, eosinophilic fasciitis.[3][4]
Epidemiology
Accurate information regarding the epidemiology regarding the incidence of aplastic anemia is generally not available. Studies suggest the incidence is 0.6 to 6.1 cases per million population; this rate is largely based on data from retrospective reviews of death registries.[5][6]
The male-to-female ratio is approximately 1:1. Although aplastic anemia occurs in all age groups, a small peak in the incidence is observed in childhood. A second peak is found in the 20 to 25-year-old age group.
Pathophysiology
Two interrelated explanations exist for aplastic anemia: extrinsic immune-mediated suppression of hematopoietic stem cells and intrinsic abnormality of marrow progenitors.[7][8]
Damaged hematopoietic stem cells mature into self-reactive T-helper cells (T1) that release cytokines interferon-? (IFN?) and tumor necrosis factor (TNF) to propagate a cytotoxic cascade to kill and suppress other hematopoietic stem cells. The exact antigens T1 cells target are unclear, but one appears to be the glucose phosphate inositol (GPI)-linked protein on cell membranes (the mechanism behind pancytopenia in PNH). Also, the genes for apoptosis and death pathways are upregulated. Moreover, immunosuppressive therapy targeting T-cells leads to a response in two-thirds of patients with idiopathic aplastic anemia, and patients with graft-versus-host disease develop aplasia in the setting of healthy bone marrow progenitors.
In the second theory, stem cells with inherent defects lose the capacity to differentiate and proliferative. Their inability to dedifferentiate can lead to clonal evolution into hematologic neoplasms, for example, myelodysplastic syndrome. This is common in patients with Fanconi anemia. Partial defects in telomeres, the component of DNA intertwined with cell division, lead to premature hematopoietic stem cell exhaustion and marrow aplasia as well. Shortened telomeres are present in cells of half the patients with aplastic anemia.
Histopathology
Bone marrow biopsy from patients with aplastic anemia will be markedly hypocellular. Fat cells and fibrotic stroma replace normal bone marrow tissue. Stray lymphocytes and plasma cells remain, the remainder is devoid of marrow progenitors.
History and Physical
Aplastic anemia presents at any age with equal distribution among gender and race. Symptoms related to the absent cell lineage (anemia, progressive weakness, pallor, and dyspnea; neutropenia, frequent and persistent minor infections, or sudden onset febrile illness; thrombocytopenia, ecchymoses, mucosal bleeding, and petechiae). Splenomegaly is not seen, and its presence suggests an alternative diagnosis. Labs will demonstrate macrocytic normochromic anemia with reticulopodia, neutropenia, and thrombocytopenia. There must be no cytologic abnormalities as this would suggest an underlying hematologic process.
Evaluation
The diagnostic criteria for aplastic anemia are the following: the presence of bone marrow hypocellularity and 2 or more cytopenias (reticulopodia less than 1% or less than 40,000/microliter, neutropenia less than 500/microliter, or thrombocytopenia less than 20,000/microliter). The moderate disease has less than 30% bone marrow cellularity; the severe disease has less than 25% cellularity or less than 50% cellularity containing fewer than 30% hematopoietic cells, and very severe meets severe criteria plus neutropenia less than 200/µL. Aspirate of bone marrow has little yield (“dry tap”). Bone marrow biopsy is essential: it will be markedly hypocellular and devoid of marrow progenitors. Genetic testing with flow cytometry and fluorescence in situ hybridization (FISH) is useful to exclude hematologic malignancies responsible for pancytopenia. Additional testing depends on the underlying condition responsible for bone marrow failure, for example, telomerase mutation testing for dyskeratosis congenital.[9][10]
Treatment / Management
Management of aplastic anemia is directed at the underlying cause. Remove the offending agent(s), if possible. Some drugs have been discontinued in the United States due to their associations with aplastic anemia (e.g., Ticlopidine, a platelet aggregation inhibitor used as primary/secondary stroke prevention or dual antiplatelet therapy following percutaneous coronary intervention; phenylbutazone, a NSAID used as an analgesic and antipyretic). Aplastic anemia associated with pregnancy is self-limited and ends with delivery. Patients with thymoma usually have full bone marrow recovery following thymectomy.[2][11][12](B3)
For patients in whom no reversible cause is found, treatment depends on age, disease severity, donor availability, and performance status. Young patients (younger than 50 years) in good health with severe disease should undergo allogeneic hematopoietic cell transplant (HCT) before initial immunosuppressive therapy. Older patients (50 years or older) in good health and young patients without an HCT donor receive full-dose immunosuppressive therapy using eltrombopag, horse/rabbit anti-thymocyte globulin (ATG), cyclosporine A, and prednisone. This combination can be tailored to single-agent eltrombopag, ATG, or cyclosporine A for less healthy individuals. Eltrombopag is a thrombopoietin non-peptide agonist that increases platelet counts and activates intracellular signal transduction pathways to increase proliferation and differentiation of marrow progenitor cells. ATG eliminates antigen-reactive T-lymphocytes and induces hematologic responses in aplastic anemia. Cyclosporine A inhibits the production and release of interleukin-II (IL-2) and inhibits IL-2 induced activation of resting T-lymphocytes. Prednisone induces cell death of immature lymphocytes. Clinical studies are underway for alternative therapies used as second-line agents.
Supportive care includes infection prophylaxis/treatment and transfusions (leukoreduced red blood cells for Hb less than 7 mg/dL or platelets less than 10,000/microliters or less than 50,000/microliters for active blood loss). Monitor for secondary hemochromatosis and administer iron chelators as indicated. Use of growth factors such as erythropoietin or granulocyte colony-stimulating factors is not recommended because there are inadequate precursor cells to generate sufficient responses.
Survival in aplastic anemia depends largely on age, disease severity, and response to initial therapy. Those who recover following drug cessation or treatment of underlying condition have a stable clinical course, as well as those with self-limited processes. Five-year survival is greater than 75% for patients who undergo bone marrow transplant from a suitable donor. The majority of untreated patients die within one year from disease-related complications (e.g., bleeding, infections, or transformation to lymphoproliferative disorders).
Differential Diagnosis
Pancytopenia can occur due to myelophthistic syndrome, a pathologic process that replaces normal bone marrow. Etiologies are solid tumor metastases (ex. lung, breast, and prostate malignancies), lymphoid or myeloid neoplasms (ex. acute myelogenous leukemia), myelofibrosis, hemophagocytic lymphohistiocytosis, osteopetrosis, or Gaucher disease. The bone marrow biopsy will not be hypocellular and reflect the underlying disease.
Isolated failure of single hematopoietic lineage is common (ex. agranulocytosis, pure red cell aplasia). These share the same causes for aplastic anemia (ex. propylthiouracil and agranulocytosis, thymoma and pure red cell aplasia). Patients will have symptoms related to the cell line involved, not all three.
Prognosis
Survival in aplastic anemia depends largely on age, disease severity, and response to initial therapy. Those who recover following drug cessation or treatment of underlying condition have stable clinical courses, as well as those with self-limited processes. Five-year survival is >75% for patients who undergo bone marrow transplant from a suitable donor. The majority of untreated patients die within one year from disease-related complications (ex. bleeding, infections, or transformation to lymphoproliferative disorders).
Complications
The most common complications of aplastic anemia include bleeding, infections, or transformation to lymphoproliferative disorders. These are managed by surveillance and symptomatic treatment including antibiotics, chemotherapy, and/or transfusions.
Deterrence and Patient Education
Aplastic anemia is a condition in which the body is unable to make blood cells that perform vital functions including infection control, oxygen transport, and tissue repair following injury. While there are many causes for this disease, many patients never find the underlying issue. Recovery is excellent for patients with identifiable causes or disease that resolves spontaneously. Patients can opt for bone marrow transplant and additional medications to provide blood products to the body. Monitor for disease complications such as bleeding, cancers or infections, and notify physicians of any changes.
Pearls and Other Issues
Pancytopenia can occur due to the myelophthisic syndrome, a pathologic process that replaces normal bone marrow. Etiologies are solid tumor metastases (e.g., lung, breast, and prostate malignancies), lymphoid or myeloid neoplasms (e.g., acute myelogenous leukemia), myelofibrosis, hemophagocytic lymphohistiocytosis, osteopetrosis, or Gaucher disease. The bone marrow biopsy will not be hypocellular and reflect the underlying disease.
Isolated failure of a single hematopoietic lineage is common (agranulocytosis, pure red cell aplasia). These share the same causes for aplastic anemia (e.g., propylthiouracil and agranulocytosis, thymoma and pure red cell aplasia). Patients will have symptoms related to the cell line involved, not all three.
Enhancing Healthcare Team Outcomes
The management of patients with aplastic anemia is an interprofessional team endeavor. The disorder can affect many organ systems and besides the disease itself, many complications can result occur following immunosuppressive therapy and hematopoietic cell transplantation. These patients need close monitoring for infections and bleeding. Because of the neutropenia, the diet has to be tailored made and should exclude dairy products, raw meat and most vegetables and fruits because of colonization by a number of microorganisms. All patients should avoid intense physical activity because they are at a risk for bleeding. Premenopausal women can develop heavy periods and should be advised to be on hormonal therapy. Finally, all patients should be educated on the need to maintain good hand and personal hygiene because they are at a very high risk for infections. [13][14](Level V)
Outcomes
Over the past 3 decades, the prognosis for patients with aplastic anemia has markedly improved because of better treatment and supportive measures. Depending on the cause, with treatment 10-year survival of 65-75 have been reported with immunosuppressive and hematopoietic cell transplantation. In fact, for matched siblings, the outcomes of hematopoietic cell transplantations are excellent. With the use of immunosuppressive therapy, there is always a risk of relapse and late clonal disease. Some data show that in patients managed with immunosuppression, telomere length of leucocytes is associated with low survival, clonal evolution, and risk of relapse. The two major causes of death in aplastic anemia include bleeding and infection. Patients who undergo cell transplantation are at risk for graft versus host disease and graft failure. [15][16](Level V)
Media
(Click Image to Enlarge)
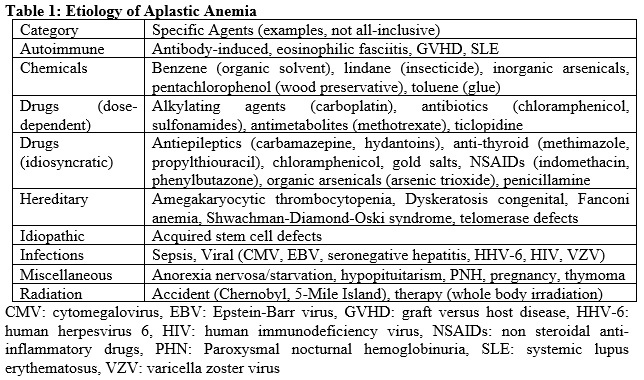
Table for causes of Aplastic Anemia Contributed by Christine A Moore
(Click Image to Enlarge)
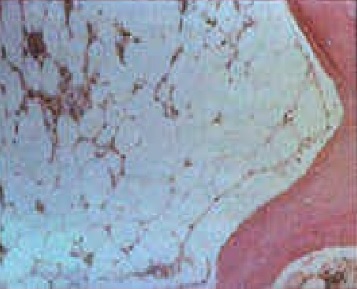
Aplastic anemia bone marrow Contributed by Ruozhi Xiao
References
Ding SX, Chen T, Wang T, Liu CY, Lu WL, Fu R. The Risk of Clonal Evolution of Granulocyte Colony-Stimulating Factor for Acquired Aplastic Anemia: A Systematic Review and Meta-Analysis. Acta haematologica. 2018:140(3):141-145. doi: 10.1159/000491816. Epub 2018 Sep 25 [PubMed PMID: 30253387]
Level 1 (high-level) evidenceGeorges GE, Doney K, Storb R. Severe aplastic anemia: allogeneic bone marrow transplantation as first-line treatment. Blood advances. 2018 Aug 14:2(15):2020-2028. doi: 10.1182/bloodadvances.2018021162. Epub [PubMed PMID: 30108110]
Level 3 (low-level) evidenceShallis RM, Ahmad R, Zeidan AM. Aplastic anemia: Etiology, molecular pathogenesis, and emerging concepts. European journal of haematology. 2018 Dec:101(6):711-720. doi: 10.1111/ejh.13153. Epub 2018 Oct 10 [PubMed PMID: 30055055]
Gadalla SM, Aubert G, Wang T, Haagenson M, Spellman SR, Wang L, Katki HA, Savage SA, Lee SJ. Donor telomere length and causes of death after unrelated hematopoietic cell transplantation in patients with marrow failure. Blood. 2018 May 24:131(21):2393-2398. doi: 10.1182/blood-2017-10-812735. Epub 2018 Apr 9 [PubMed PMID: 29632022]
Li SS, Hsu YT, Chang C, Lee SC, Yen CC, Cheng CN, Chen JS, Lin SH, Chang KC, Chen TY. Incidence and treatment outcome of aplastic anemia in Taiwan-real-world data from single-institute experience and a nationwide population-based database. Annals of hematology. 2019 Jan:98(1):29-39. doi: 10.1007/s00277-018-3486-3. Epub 2018 Sep 3 [PubMed PMID: 30178191]
Vaht K, Göransson M, Carlson K, Isaksson C, Lenhoff S, Sandstedt A, Uggla B, Winiarski J, Ljungman P, Brune M, Andersson PO. Incidence and outcome of acquired aplastic anemia: real-world data from patients diagnosed in Sweden from 2000-2011. Haematologica. 2017 Oct:102(10):1683-1690. doi: 10.3324/haematol.2017.169862. Epub 2017 Jul 27 [PubMed PMID: 28751565]
Schoettler ML, Nathan DG. The Pathophysiology of Acquired Aplastic Anemia: Current Concepts Revisited. Hematology/oncology clinics of North America. 2018 Aug:32(4):581-594. doi: 10.1016/j.hoc.2018.03.001. Epub 2018 May 8 [PubMed PMID: 30047412]
Yamazaki H. [Acquired aplastic anemia: recent advances in pathophysiology and treatment]. [Rinsho ketsueki] The Japanese journal of clinical hematology. 2018:59(6):711-715. doi: 10.11406/rinketsu.59.711. Epub [PubMed PMID: 29973449]
Level 3 (low-level) evidenceGnanaraj J, Parnes A, Francis CW, Go RS, Takemoto CM, Hashmi SK. Approach to pancytopenia: Diagnostic algorithm for clinical hematologists. Blood reviews. 2018 Sep:32(5):361-367. doi: 10.1016/j.blre.2018.03.001. Epub 2018 Mar 5 [PubMed PMID: 29555368]
Bluteau O, Sebert M, Leblanc T, Peffault de Latour R, Quentin S, Lainey E, Hernandez L, Dalle JH, Sicre de Fontbrune F, Lengline E, Itzykson R, Clappier E, Boissel N, Vasquez N, Da Costa M, Masliah-Planchon J, Cuccuini W, Raimbault A, De Jaegere L, Adès L, Fenaux P, Maury S, Schmitt C, Muller M, Domenech C, Blin N, Bruno B, Pellier I, Hunault M, Blanche S, Petit A, Leverger G, Michel G, Bertrand Y, Baruchel A, Socié G, Soulier J. A landscape of germ line mutations in a cohort of inherited bone marrow failure patients. Blood. 2018 Feb 15:131(7):717-732. doi: 10.1182/blood-2017-09-806489. Epub 2017 Nov 16 [PubMed PMID: 29146883]
Yoshida N, Kojima S. Updated Guidelines for the Treatment of Acquired Aplastic Anemia in Children. Current oncology reports. 2018 Jun 30:20(9):67. doi: 10.1007/s11912-018-0716-8. Epub 2018 Jun 30 [PubMed PMID: 29961134]
Samarasinghe S, Veys P, Vora A, Wynn R. Paediatric amendment to adult BSH Guidelines for aplastic anaemia. British journal of haematology. 2018 Jan:180(2):201-205. doi: 10.1111/bjh.15066. Epub 2017 Dec 28 [PubMed PMID: 29285764]
Zakaria Z, Kaliaperumal C, Crimmins D, Caird J. Neurosurgical management in children with bleeding diathesis: auditing neurological outcome. Journal of neurosurgery. Pediatrics. 2018 Jan:21(1):38-43. doi: 10.3171/2017.6.PEDS16574. Epub 2017 Nov 10 [PubMed PMID: 29125443]
Dietz AC, Mehta PA, Vlachos A, Savage SA, Bresters D, Tolar J, Boulad F, Dalle JH, Bonfim C, de la Fuente J, Duncan CN, Baker KS, Pulsipher MA, Lipton JM, Wagner JE, Alter BP. Current Knowledge and Priorities for Future Research in Late Effects after Hematopoietic Cell Transplantation for Inherited Bone Marrow Failure Syndromes: Consensus Statement from the Second Pediatric Blood and Marrow Transplant Consortium International Conference on Late Effects after Pediatric Hematopoietic Cell Transplantation. Biology of blood and marrow transplantation : journal of the American Society for Blood and Marrow Transplantation. 2017 May:23(5):726-735. doi: 10.1016/j.bbmt.2017.01.075. Epub 2017 Jan 20 [PubMed PMID: 28115275]
Level 3 (low-level) evidenceRice C, Eikema DJ, Marsh JCW, Knol C, Hebert K, Putter H, Peterson E, Deeg HJ, Halkes S, Pidala J, Anderlini P, Tischer J, Kroger N, McDonald A, Antin JH, Schaap NP, Hallek M, Einsele H, Mathews V, Kapoor N, Boelens JJ, Mufti GJ, Potter V, Pefault de la Tour R, Eapen M, Dufour C. Allogeneic Hematopoietic Cell Transplantation in Patients Aged 50Years or Older with Severe Aplastic Anemia. Biology of blood and marrow transplantation : journal of the American Society for Blood and Marrow Transplantation. 2019 Mar:25(3):488-495. doi: 10.1016/j.bbmt.2018.08.029. Epub 2018 Sep 5 [PubMed PMID: 30194027]
Scheinberg P. Recent Advances and Long-Term Results of Medical Treatment of Acquired Aplastic Anemia: Are Patients Cured? Hematology/oncology clinics of North America. 2018 Aug:32(4):609-618. doi: 10.1016/j.hoc.2018.03.003. Epub 2018 May 18 [PubMed PMID: 30047414]
Level 3 (low-level) evidence