Introduction
Polycystic liver disease (PLD) is a rare genetic disorder characterized by mutations in genes encoding for proteins involved in the transport of fluid and growth of epithelial cells in the liver (see Image. Polycystic Liver Disease).[1] These mutations lead to the replacement of normal liver tissue with fluid-filled liver cysts. There are two distinct forms of PLD; PLD in isolation and PLD in association with polycystic kidney disease (PKD). The majority of patients with PLD are asymptomatic and diagnosed incidentally on imaging.[2] However, in a small percentage of patients, hepatomegaly can lead to abdominal pain, distension, and compression of adjacent organs, potentially affecting the quality of life.[3][1] PLD is diagnosable using ultrasonography, computed tomography (CT) scan, or magnetic resonance imaging (MRI). For the patient with symptomatic PLD, the main goal is to decrease liver volume. There are currently several surgical options available, including cyst fenestration, hepatic resection, and liver transplant. There are also medical therapies now under investigation.[4] This activity discusses the diagnosis and management of PLD.
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
The cause of PLD is the result of mutations in several genes. In isolated PLD, germline mutations occur in the PRKCSH, and SEC63 genes are in charge of making the proteins sec-63 and hepatocystin and are responsible for fluid transportation and epithelial cell growth.[4][3] They are expressed on hepatocytes and cholangiocytes, although the underlying mechanism of how they are involved in cyst formation remains unclear. The second group of PLD is those associated with PKD. There are two forms of PKD; autosomal-dominant PKD (ADPKD) and autosomal-recessive PKD (ARPKD). In this form of PLD, the kidney has the greatest involvement. Patients tend to develop renal complications such as renal failure, with most requiring dialysis and kidney transplant. The two genes believed to be the culprit of cyst formation in ADPKD are PKD1 and PKD2.[1] These genes are responsible for making polycystin 1 and polycystin 2, regulating fluid secretion and epithelial cell growth. The mutation of these genes leads to dysregulation of fluid secretion and abnormal growth, ultimately leading to cyst formation.[2] Most patients with ARPKD, on the other hand, die shortly after birth due to pulmonary complications. Those that do survive tend to develop liver fibrosis rather than cysts.[5]
Epidemiology
For years, the belief was that the only way PLD could develop was in patients with PKD. Clinically, isolated PLD was identified as a separate condition in the 1950s, and it received genetic confirmation in 2003, following the analysis of eight Finnish families.[6] Isolated PLD has a prevalence in the general population of 1 to 10 cases per 1000000 individuals, while ADPKD ranged from 1 in 400 to 1 in 1000. ADPKD represents roughly 80 to 90% of all patients with PLD.[4][2] Due to the autosomal inheritance pattern of Isolated PLD, males and females should be at equal risk. However, findings show that the female to male ratio is 6 to 1; this is due to higher estrogen levels in females compared to males. Estrogen has been shown to stimulate cyst production, resulting in females being more symptomatic and having more extensive experiences with PLD than males.[4]
Pathophysiology
Isolated PLD results from embryonic ductal plate malformation of the intrahepatic bile ducts and cilia of the cholangiocytes. In normal bile duct formation, there is a complex sequence of events that involves both growth and apoptosis. In patients with isolated PLD, there is a disruption in this sequence, and no apoptosis occurs. As a result, complexes of disconnected intralobular bile ductules known as “Von Meyenburg complexes” form. Cysts arise from these complexes and continue to grow with time.[1][4] Cilia play an essential role in the production of cholangiocytes in the liver. Cilia can modulate intracellular levels of adenosine 3',5'-cyclic monophosphate (cAMP)/calcium, detect osmolarity changes in the bile, and have mechanosensory capabilities. Defects in cilia lead to increased levels of calcium, which increases cAMP. The increase in cAMP drives the hyperproduction of cholangiocytes and the formation of cysts. The increase in cAMP also alters the fluid balance in the lumen of the biliary ducts.[1]
Histopathology
Liver cysts originate from medium-sized bile ducts. On histopathology, liver biopsy showed multiple diffuse cystic lesions resembling solitary cysts, lined by cuboidal to flat biliary epithelium surrounded by fibrous stroma, containing straw-colored fluid.[7][8] About 40% of these patients will have identifiable von Meyenburg complexes, and they do not contain pigmented material.
History and Physical
Most patients with isolated PLD are asymptomatic, and estimates are that 80% of patients are usually diagnosed incidentally on imaging studies. Symptomatic severity is dependent on the number, size, and location of the cysts. In symptomatic patients, the cysts are impeding on nearby intrahepatic structures such as the inferior vena cava, hepatic veins, portal veins, or bile ducts. The common symptoms in patients with PLD include abdominal distension, shortness of breath, postprandial fullness, esophageal reflux, abdominal pain, and back pain. If patients have reflux or early satiety/postprandial fullness, they can be at risk for malnutrition and failure to thrive.
Compression of nearby vascular structures can lead to portal hypertension, and patients may present with variceal bleeding, jaundice, ascites, and/or encephalopathy.[2][3][9] Portal hypertension can be caused by a reduction in hepatic vein outflow or by compression of portal vein inflow. Signs and symptoms consistent with portal vein inflow obstruction are abdominal pain, hepatomegaly, and transudative ascites; this can result from direct compression from the cysts or the development of hepatic vein thrombosis from stasis.
Liver cysts can also cause a Budd-Chiari-like syndrome if venous drainage from the liver is obstructed and may present with the classic triad of abdominal pain, ascites, and hepatomegaly.[4][2][3] Patients who do develop advanced liver disease from isolated PLD are at risk for complications such as infections, torsion, rupture, and hemorrhage of the cysts.[1][10] End-stage liver disease or cirrhosis can develop with extremely increased liver volumes. Patients with severe PLD usually do not meet the Model for End-Stage Liver Disease (MELD) criteria. However, they can obtain transplants with exception points for failure to thrive and malnutrition.[2]
Evaluation
The diagnosis of PLD is made with imaging studies. Unfortunately, at this time, there are no unified radiologic diagnostic criteria. Most individuals are diagnosed in the fourth to fifth decades of life. Ultrasound imaging will show hyperechoic areas in the subcapsular portion of the liver. MRI and CT imaging is another alternative, and on non-contrast images, the cyst will appear hypodense and well circumscribed.[1]
Reynolds criteria for PLD are typically used for diagnosis. This takes into account family history, age, and liver phenotype. An ultrasound to identify cysts in the liver and kidney is recommended as the initial test. ADPKD should be excluded before making the diagnosis of isolated PLD, and the unified Ravine criteria for ADPKD diagnosis can be used to make this exclusion. Unfortunately, up to one-third of patients with isolated PLD can have a few kidney cysts, making it difficult to distinguish between the two. In patients with renal cysts and kidney phenotype, the diagnosis of ADPKD is made for patients aged 15 to 39 years with the presence of three or more renal cysts, two or more renal cysts for patients aged 40 to 59 years, and four or more renal cysts for patients over the age of 60.[2]
Some studies propose that sporadic cases of isolated PLD can be diagnosed when a patient has 15 to 20 cysts and no family history. However, based on Reynold’s criteria, patients need a positive family history and exclusion of ADPKD. If these two criteria are met, they are subdivided by age and liver phenotype. Patients under the age of 40 and with one or more hepatic cysts or patients over the age of 40 and with four or more hepatic cysts meet the criteria for isolated PLD.[4][2]
A laboratory diagnostic test does not yet exist for pure isolated PLD. Most patients have normal synthetic liver function and, on occasion, can have mild elevations of gamma-glutamyl transferase (GGT) and alkaline phosphatase. On the other hand, patients with ADPKD have signs of renal dysfunction and variations in their creatinine, blood urea nitrogen (BUN), and/or glomerular filtration rate (GFR) levels.[4]
Treatment / Management
Most patients are asymptomatic, and no treatment or management is necessary for patients with isolated PLD. For symptomatic patients, the principal aim of treatment is to reduce symptoms by decreasing cyst volume and liver size. These interventions can divide into three broad categories: medical therapy, surgical therapy, and liver transplant as a last resort.
Medical Therapy
There is a lack of adequate medical treatment options available for patients with PLD and are currently under investigation in clinical trials.[3]
Somatostatin Receptor Antagonist
Octreotide is a somatostatin receptor antagonist that has been shown to affect patients with PLD positively. cAMP plays a vital role in the development of cell proliferation and fluid secretion. Octreotide inhibits cAMP in cystic cholangiocytes and therefore leads to decreased fluid production and cell proliferation. Clinical trials have demonstrated its success in reducing liver volume and improving patient symptoms.[2][11] Currents recommendations are to give long-acting somatostatin analog once every month. However, there is currently not enough data on the long-term effects of remaining on these medications. Also, the cost of this therapy prohibits many patients from pursuing it as a treatment. It requires evidence of poor quality of life from moderate to severe disease to have the drug approved for treatment. Nevertheless, these medications are well tolerated, and the side effects, which include diarrhea, abdominal discomfort, and gallstones, are usually transient.[4](B3)
Mammalian Target of Rapamycin (mTOR) Inhibitor
mTOR is inappropriately activated in patients with PLD and is thought to be responsible for cystic epithelium proliferation. Also, it increases the synthesis and secretion of vascular endothelial growth factor (VEGF), leading to the growth of cystic epithelium. The mTOR inhibitor sirolimus has been demonstrated to block the disordered, unregulated proliferative response of polycystic epithelial cells.[3] In a retrospective study on kidney transplant patients by Qian et al.[12] showed sirolimus was associated with an 11.9 +/- 0.03% reduction in polycystic liver volume. In contrast, treatment with tacrolimus correlated with a 14.1 +/- 0.09% increase. While the recently published two randomized controlled trials on the patients with ADPKD showed the opposite result of mTOR inhibitors. Walz et al. showed mTOR inhibitor (everolimus) appeared to slow the total kidney volume increase without improvement in the estimated GFR.[13] Serra et al. showed that treatment with sirolimus for 18 months did not slow kidney growth.[14] At present, no substantial evidence suggests the routine use of sirolimus in patients with PLD, and these require confirmation in larger randomized controlled trials. (A1)
Vasopressin-2-receptor Antagonists
Vasopressin-2 receptor, when stimulated, increases cAMP production and can lead to the growth and formation of cysts. Several studies have shown that using antagonists to block this receptor can slow down cyst formation and growth. One of the larger studies completed by Torres et al.[15] looked at tolvaptan use in 1445 patients with ADPKD over three years slowed the growth rate and increased total kidney volume. The study did include patients with liver involvement, but they did not analyze tolvaptan’s effect on liver cysts. It is hypothesized that vasopressin-2-receptor antagonists would have little effect on hepatic cyst epithelium because of the lack of the vasopressin-2 receptor on these cells. Despite this, there have been isolated case studies that suggest that tolvaptan may reduce liver volume and improve abdominal symptoms. More extensive studies are necessary on patients with PLD in the future.[15](A1)
Estrogen Receptor Antagonists
The current belief is that estrogen plays a role in cyst growth, and as a result, the disease tends to affect women more than men. However, there have been no formal studies conducted besides anecdotal reports that look at the effects of estrogen receptor antagonists in both forms of PLD.[3]
Surgical Therapy
Percutaneous Cyst Aspiration and Sclerotherapy
It involves aspiration of a cyst under ultrasound or CT guidance followed by injection of a sclerosing agent that leads to the destruction of the epithelial lining, inhibiting fluid production.[16][17] It is mainly indicated for a large symptomatic liver cyst that is likely to be responsible for the symptoms. The cysts with a diameter of >5 cm are good candidates for therapy. The most commonly used sclerosing agent includes ethanol, tetracycline, and minocycline.[9] A recent systemic review by Wijnands et al.[17] showed excellent results of efficacy and safety of aspiration and sclerotherapy. The analysis included 16 studies, which included 526 patients with a total of 588 treated cysts. Proportional cyst volume reduction ranged between 76% and 100% after a median follow-up period of 1 to 54 months, 72% to 100% of patients reported symptom reduction, and 56 to 100% reported the disappearance of symptoms. Postprocedural pain is more common with rate, at a rate of 5% to 90% among studies.(A1)
Laparoscopic Cyst Fenestration
It involves aspiration and deroofing of the cyst in a single procedure. The main advantage of the procedure that multiple cysts are treatable at the same time. Indications for this procedure are when there are a few large dominant cysts in the anterior segments of the right lobe or the left lateral segments. The main complications include post-procedure drainage, ascites, pleural effusion, arterial or venous bleeding, and bile leaks.[3][9] A recent systemic systematic review and meta-analysis by Bernts et al. evaluated clinical response after laparoscopic fenestration of symptomatic hepatic cysts.[18] The analysis included 62 studies with a total of 1314 patients. Symptomatic relief and symptomatic recurrence after laparoscopic fenestration were 90.2% (95% CI 84.3–94.9) and 9.6% (95% CI 6.9–12.8), respectively. Postoperative complications occurred in 10.8% (95% CI 8.1–13.9) with procedure-related mortality was 1.0% (95% CI 0.5–1.6). In a subgroup analysis, symptomatic recurrence rate and risk of complications are significantly higher in patients with PLD.(A1)
Segmental Hepatic Resection
Segmental hepatic resection merits consideration in patients whose cysts are clustered to individual segments but have enough remnant liver with normal liver parenchyma. The procedure is usually only for patients with massive hepatomegaly with severe symptoms and when liver transplantation is unwarranted.[9] At least 25% to 30% of the post-resection normal liver parenchyma is recommended for a good outcome.[19] The main complications include venous bleed or bile leaks due to distortion of the intrahepatic vasculature and biliary system by cysts.[4][3] Also, there is an increased risk of developing adhesions that can complicate future liver transplantation if required in the future. (B3)
Liver Transplantation
Liver transplantation is generally only for symptomatic patients where resection is not a feasible option due to diffuse cystic disease; this is also recommended in the rare scenario where there is impaired hepatic function. The current guidelines for liver transplant rely on the Model for End-Stage Liver Disease (MELD). Patients with PLD have low MELDs due to the preserved synthetic function of the liver. They can, however, achieve exception points if there is evidence of malnutrition and failure to thrive.
Patients are proposed for transplant if they have high symptomatic PLD, have failed alternative interventions, and have severe malnutrition. Severe malnutrition is defined as a serum albumin level less than 2.2 g/dL, or mid-arm circumference in the non-dominant arm of less than 23.1 cm in a female and less than 23.8 cm in a male. This patient will receive an initial MELD of 15 if their creatinine clearance is less than 30 mL/minute or a score of 20 if the creatinine clearance is less than 30 mL/min. Patients with ADPKD should consider evaluation for both a liver and kidney transplant. Patients receiving a liver transplant have a five-year graft survival rate of 87.5% and an overall survival rate of 92.3%.[4]
Differential Diagnosis
When considering PLD, other potential causes should be ruled out. These include neoplasm, infections, and trauma. Imaging allows the distinguishing of PLD from hepatic metastases or an abscess. Other communicative biliary tree disorders to consider would be Caroli disease and Caroli syndrome. These two diseases are congenital genetic disorders of the intrahepatic bile ducts. These are typically seen in childhood, while isolated PLD occurs in adulthood. They are also associated with congenital hepatic fibrosis.[3]
Prognosis
PLD is a progressive disease, and only a small subset of patients develop severe symptoms. Supportive management with as-needed analgesics is the first-line treatment in PLD patients with acute or chronic abdominal pain. The primary aim of PLD treatment is focused on the reduction in liver volume to improve symptoms and quality of life.[9][1] The different invasive procedures that have shown improved outcomes include aspiration sclerotherapy, laparoscopic cyst fenestration, liver resection, or liver transplantation.[9] Although these procedures carry the risk of significant morbidity and potential benefits should be weighed carefully against the complications of procedures.
Complications
Several complications are notable with PLD patients. These complications can be broken down into intra-cystic complications and liver volume complications.
Intra-cystic Complications
Hepatic cyst hemorrhage
It typically manifests as acute right upper quadrant abdominal pain that progresses in the first few days and resolves spontaneously.[1] The etiology is thought to be high intracystic pressure, rapid growth, and direct trauma. Diagnosis is typically seen on ultrasound imaging, while CT or MRI imaging with contrast may be required to distinguish hemorrhage from malignancy.[20] Cystadenocarcinoma, cystic adenoma, and hemorrhagic cysts all appear similar on ultrasound imaging.[2]
Hepatic Cyst Infections
Hepatic cyst infections are rare (1%) complications in patients with PLD and are believed to result from the translocation of bacteria from the intestines. It is typically present as right upper quadrant pain, fever, and malaise.[1] The imaging studies showing the hepatic cyst wall thickening and complex fluid are suggestive for infection but not accurate. The cyst aspiration showing the presence of inflammatory cells and bacteria is the gold standard for diagnosis. The two most common bacteria that cause these infections are Escherichia coli and Klebsiella species. A fluorodeoxyglucose-positron emission tomography (FDG-PET) can be used to identify the infected cysts, as the epithelia of these cysts will accumulate the FDG. Unfortunately, it is not the gold standard, as the accuracy only seems to improve in the later stages of the infection.[21][22] Extremely high carbohydrate antigen (CA) 19-9 levels were found in patients with hepatic cyst infection and declining during recovery.[23] Treatment is usually broad-spectrum antibiotics, and therapy is narrowed down based on culture results. There is a 64% failure rate with just antibiotic treatment, and in these cases, cyst drainage is recommended alongside the antibiotics. C-reactive protein has also been found to be useful in monitoring treatment response.[2]
Hepatic Cyst Ruptures
Hepatic cyst rupture is an extremely rare complication and has only been published in case reports.[24] It occurs due to an increase in cyst volume that can occur spontaneously or secondary to hemorrhage. Imaging studies will show free fluid around the liver as well as a residual cyst. Patients will typically have severe abdominal pain. In most cases, treatment is conservative management and supportive care. However, if there is hemodynamic instability, treatment consists of drainage of ascites or liver cysts or surgical evaluation and intervention.[2]
Liver Volume Complications
Liver volume-related complications are another rare occurrence. This refers to complications arising from the cyst putting pressure on adjacent organs or by an enlarged liver. This has only been reported in case reports. Patients can develop obstructive jaundice, portal vein occlusion, portal hypertension with splenic varices, Budd-Chiari syndrome, and compression of the inferior vena cava, leading to edema and ascites. Treatments are individualized and based on the complications.[2]
The last rare complication that can develop in patients with isolated PLD is cystic carcinoma. This typically presents with abdominal pain and weight loss. The patient usually has the advanced liver disease at this point.[25]
Deterrence and Patient Education
PLD is a progressive disease, and only a small subset of patients develop severe symptoms. Supportive management is recommended with a patient with mild symptoms, while the patient with severe symptoms can be managed surgically, with liver transplantation being the only curative option. There is a lack of effective medical treatment options available for patients with PLD and are currently under investigation in clinical trials.[1][3] The risk factors for the development of severe PLD are age, female sex, prior use of exogenous estrogens, and multiple pregnancies.[26] PLD has an autosomal dominant inheritance pattern, and the recurrence risk is 50%. Thus, genetic counseling is advised in patients severely affected by PLD that may allow differentiation between ADPKD and isolated PLD.[27][28] The screening for PLD in the general population is not recommended since the prevalence of the disease is relatively low, and it will be of low yield.
Enhancing Healthcare Team Outcomes
Enhancing outcomes in symptomatic patients with PLD requires an interprofessional team approach from primary care providers, hospitalists, hepatologists, specialty-trained nurses, pharmacists, and interventional radiologists and surgeons at times, all collaborating across disciplines to achieve optimal patient results. Clinicians and other specialists will manage the condition overall, but medications should be vetted through a board-certified pharmacotherapy pharmacist, who can collaborate on agent selection, verify dosing, and oversee the medication regimen for drug interactions. Specialized nursing can administer medications, monitor treatment effectiveness, and alert clinicians to any possible adverse effects. In surgical cases, nursing will have involvement throughout the process from surgical prep, assisting during the procedure, and post-operative care, informing the clinician of any issues that they encounter. Such an interprofessional team of health professionals providing an integrated approach to the care of these patients can help to achieve the best possible outcomes. [Level 5]
Media
(Click Image to Enlarge)
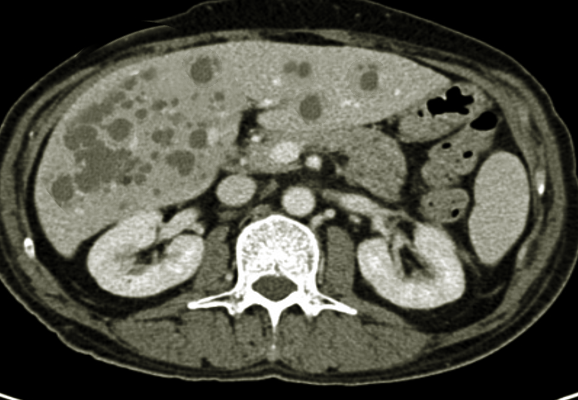
Polycystic Liver Disease
Contributed by S Bhimji, MD
References
Cnossen WR, Drenth JP. Polycystic liver disease: an overview of pathogenesis, clinical manifestations and management. Orphanet journal of rare diseases. 2014 May 1:9():69. doi: 10.1186/1750-1172-9-69. Epub 2014 May 1 [PubMed PMID: 24886261]
Level 3 (low-level) evidencevan Aerts RMM, van de Laarschot LFM, Banales JM, Drenth JPH. Clinical management of polycystic liver disease. Journal of hepatology. 2018 Apr:68(4):827-837. doi: 10.1016/j.jhep.2017.11.024. Epub 2017 Nov 24 [PubMed PMID: 29175241]
Everson GT. Polycystic liver disease. Gastroenterology & hepatology. 2008 Mar:4(3):179-81 [PubMed PMID: 21904493]
Masyuk TV, Masyuk AI, LaRusso NF. Therapeutic Targets in Polycystic Liver Disease. Current drug targets. 2017:18(8):950-957. doi: 10.2174/1389450116666150427161743. Epub [PubMed PMID: 25915482]
Torres VE. Treatment of polycystic liver disease: one size does not fit all. American journal of kidney diseases : the official journal of the National Kidney Foundation. 2007 Jun:49(6):725-8 [PubMed PMID: 17533013]
Tahvanainen P, Tahvanainen E, Reijonen H, Halme L, Kääriäinen H, Höckerstedt K. Polycystic liver disease is genetically heterogeneous: clinical and linkage studies in eight Finnish families. Journal of hepatology. 2003 Jan:38(1):39-43 [PubMed PMID: 12480558]
Desmet VJ. Ludwig symposium on biliary disorders--part I. Pathogenesis of ductal plate abnormalities. Mayo Clinic proceedings. 1998 Jan:73(1):80-9 [PubMed PMID: 9443684]
Gallegos M,Bradly DP,Jakate SM, Image of the month. Polycystic liver disease leading to liver failure and transplantation. Clinical gastroenterology and hepatology : the official clinical practice journal of the American Gastroenterological Association. 2010 Apr; [PubMed PMID: 19465155]
Level 3 (low-level) evidenceDrenth JP, Chrispijn M, Nagorney DM, Kamath PS, Torres VE. Medical and surgical treatment options for polycystic liver disease. Hepatology (Baltimore, Md.). 2010 Dec:52(6):2223-30. doi: 10.1002/hep.24036. Epub [PubMed PMID: 21105111]
Level 3 (low-level) evidenceMacutkiewicz C, Plastow R, Chrispijn M, Filobbos R, Ammori BA, Sherlock DJ, Drenth JP, O'Reilly DA. Complications arising in simple and polycystic liver cysts. World journal of hepatology. 2012 Dec 27:4(12):406-11. doi: 10.4254/wjh.v4.i12.406. Epub [PubMed PMID: 23355921]
Masyuk TV, Masyuk AI, Torres VE, Harris PC, Larusso NF. Octreotide inhibits hepatic cystogenesis in a rodent model of polycystic liver disease by reducing cholangiocyte adenosine 3',5'-cyclic monophosphate. Gastroenterology. 2007 Mar:132(3):1104-16 [PubMed PMID: 17383431]
Level 3 (low-level) evidenceQian Q, Du H, King BF, Kumar S, Dean PG, Cosio FG, Torres VE. Sirolimus reduces polycystic liver volume in ADPKD patients. Journal of the American Society of Nephrology : JASN. 2008 Mar:19(3):631-8. doi: 10.1681/ASN.2007050626. Epub 2008 Jan 16 [PubMed PMID: 18199797]
Level 2 (mid-level) evidenceWalz G, Budde K, Mannaa M, Nürnberger J, Wanner C, Sommerer C, Kunzendorf U, Banas B, Hörl WH, Obermüller N, Arns W, Pavenstädt H, Gaedeke J, Büchert M, May C, Gschaidmeier H, Kramer S, Eckardt KU. Everolimus in patients with autosomal dominant polycystic kidney disease. The New England journal of medicine. 2010 Aug 26:363(9):830-40. doi: 10.1056/NEJMoa1003491. Epub 2010 Jun 26 [PubMed PMID: 20581392]
Level 1 (high-level) evidenceSerra AL, Poster D, Kistler AD, Krauer F, Raina S, Young J, Rentsch KM, Spanaus KS, Senn O, Kristanto P, Scheffel H, Weishaupt D, Wüthrich RP. Sirolimus and kidney growth in autosomal dominant polycystic kidney disease. The New England journal of medicine. 2010 Aug 26:363(9):820-9. doi: 10.1056/NEJMoa0907419. Epub 2010 Jun 26 [PubMed PMID: 20581391]
Level 1 (high-level) evidenceTorres VE, Chapman AB, Devuyst O, Gansevoort RT, Grantham JJ, Higashihara E, Perrone RD, Krasa HB, Ouyang J, Czerwiec FS, TEMPO 3:4 Trial Investigators. Tolvaptan in patients with autosomal dominant polycystic kidney disease. The New England journal of medicine. 2012 Dec 20:367(25):2407-18. doi: 10.1056/NEJMoa1205511. Epub 2012 Nov 3 [PubMed PMID: 23121377]
Level 1 (high-level) evidencevan Keimpema L, de Koning DB, Strijk SP, Drenth JP. Aspiration-sclerotherapy results in effective control of liver volume in patients with liver cysts. Digestive diseases and sciences. 2008 Aug:53(8):2251-7. doi: 10.1007/s10620-007-0121-x. Epub 2008 Feb 26 [PubMed PMID: 18299984]
Wijnands TF, Görtjes AP, Gevers TJ, Jenniskens SF, Kool LJ, Potthoff A, Ronot M, Drenth JP. Efficacy and Safety of Aspiration Sclerotherapy of Simple Hepatic Cysts: A Systematic Review. AJR. American journal of roentgenology. 2017 Jan:208(1):201-207. doi: 10.2214/AJR.16.16130. Epub 2016 Nov 8 [PubMed PMID: 27824501]
Level 1 (high-level) evidenceBernts LHP, Echternach SG, Kievit W, Rosman C, Drenth JPH. Clinical response after laparoscopic fenestration of symptomatic hepatic cysts: a systematic review and meta-analysis. Surgical endoscopy. 2019 Mar:33(3):691-704. doi: 10.1007/s00464-018-6490-8. Epub 2018 Oct 17 [PubMed PMID: 30334152]
Level 1 (high-level) evidenceSchindl MJ, Redhead DN, Fearon KC, Garden OJ, Wigmore SJ, Edinburgh Liver Surgery and Transplantation Experimental Research Group (eLISTER). The value of residual liver volume as a predictor of hepatic dysfunction and infection after major liver resection. Gut. 2005 Feb:54(2):289-96 [PubMed PMID: 15647196]
Marion Y, Brevartt C, Plard L, Chiche L. Hemorrhagic liver cyst rupture: an unusual life-threatening complication of hepatic cyst and literature review. Annals of hepatology. 2013 Mar-Apr:12(2):336-9 [PubMed PMID: 23396748]
Level 3 (low-level) evidencePaschali AN, Georgakopoulos AT, Pianou NK, Anagnostopoulos CD. (18)F-fluorodeoxyglucose positron emission tomography/computed tomography in infected polycystic kidney disease. World journal of nuclear medicine. 2015 Jan-Apr:14(1):57-9. doi: 10.4103/1450-1147.150553. Epub [PubMed PMID: 25709548]
Level 3 (low-level) evidencePijl JP, Kwee TC, Slart RHJA, Glaudemans AWJM. FDG-PET/CT for diagnosis of cyst infection in autosomal dominant polycystic kidney disease. Clinical and translational imaging. 2018:6(1):61-67. doi: 10.1007/s40336-017-0261-8. Epub 2018 Feb 12 [PubMed PMID: 29568734]
Kanaan N, Goffin E, Pirson Y, Devuyst O, Hassoun Z. Carbohydrate antigen 19-9 as a diagnostic marker for hepatic cyst infection in autosomal dominant polycystic kidney disease. American journal of kidney diseases : the official journal of the National Kidney Foundation. 2010 May:55(5):916-22. doi: 10.1053/j.ajkd.2009.12.023. Epub 2010 Feb 26 [PubMed PMID: 20189277]
Level 3 (low-level) evidenceShutsha E, Brenard R. Hepatic cyst rupture after a coughing fit. Journal of hepatology. 2003 Jun:38(6):870 [PubMed PMID: 12763385]
Level 3 (low-level) evidenceEverson GT, Helmke SM, Doctor B. Advances in management of polycystic liver disease. Expert review of gastroenterology & hepatology. 2008 Aug:2(4):563-76. doi: 10.1586/17474124.2.4.563. Epub [PubMed PMID: 19072404]
Level 3 (low-level) evidenceGevers TJ, Drenth JP. Diagnosis and management of polycystic liver disease. Nature reviews. Gastroenterology & hepatology. 2013 Feb:10(2):101-8. doi: 10.1038/nrgastro.2012.254. Epub 2013 Jan 8 [PubMed PMID: 23296249]
Hildebrandt F, Benzing T, Katsanis N. Ciliopathies. The New England journal of medicine. 2011 Apr 21:364(16):1533-43. doi: 10.1056/NEJMra1010172. Epub [PubMed PMID: 21506742]
Gunay-Aygun M. Liver and kidney disease in ciliopathies. American journal of medical genetics. Part C, Seminars in medical genetics. 2009 Nov 15:151C(4):296-306. doi: 10.1002/ajmg.c.30225. Epub [PubMed PMID: 19876928]