Introduction
Congenital hereditary endothelial dystrophy (CHED) presents as bilateral corneal opacification present at birth or in the immediate newborn period and is due to an autosomal recessive mutation affecting the corneal endothelium.[1][2]
Opacification is the result of stromal edema and Descemet membrane thickening due to endothelial dysfunction.[3] The current treatment for CHED is surgical, requiring a corneal tissue graft either by penetrating keratoplasty (PK) or endothelial keratoplasty (EK).[2] Recent studies have also shown promise in treatment with NSAIDs for certain mutational variants of the disease. Delaying treatment of CHED can result in poor visual development and amblyopia.[4][5]
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
In the recent past, congenital hereditary endothelial dystrophy was classified in either of two categories: Autosomal dominant, progressive (CHED1), and Autosomal recessive, nonprogressive (CHED2). CHED1 is extremely rare, only found in five family pedigrees, and was later re-classified as a form of posterior polymorphous corneal dystrophy (PPCD) after clinical, histopathological, and electron microscopy showed more consistency with PPCD.[1][6] Separate classifications no longer exist, and CHED2 is now recognized as simply CHED, an autosomal recessive, nonprogressive disease with corneal opacification in the perinatal period.[1]
The CHED mutation is due to an affected gene, SLC4A11, on chromosome 20p13.[4] SLC4A11 codes for a transmembrane protein that acts as a bicarbonate transporter, actively countering the osmotic gradient by pumping fluid from the stroma into the aqueous humor.[1][3][4] Mutations of SLC4A11 in CHED most commonly result in a truncated BTR1 protein that is unable to reach the cellular membrane and perform appropriately.[7] Dysfunction of this protein results in stromal edema, disruption of collagen fibrils, and thickening of the Descemet’s membrane due to abnormal secretion of the endothelium culminating in corneal opacification.[3]
The gene SLC4A11 is also expressed in fibrocytes of the inner ear and renal epithelium, suggesting possible comorbidities.[8][9] The SLC4A11 gene is not strictly related to CHED, as mutations can also result in Fuchs endothelial corneal dystrophy (FECD) and Harboyan Syndrome.[10][11] A 2019 study evaluated the parents of nine CHED patients and reported that one parent of each group showed early developments of FECD due to a heterozygous SLC4A11 mutation.[10] Harboyan syndrome is characterized as CHED with accompanying progressive sensorineural hearing loss in ages 10-25 years. This is an allelic disorder, found on the same gene and locus as the CHED mutation, and identical to CHED at birth.[3] Findings have suggested that CHED developing due to homozygous mutations of SLC4A11 more often progress to Harboyan Syndrome.[12]
Studies have found that CHED is associated with mutation heterogeneity, such that a variety of mutations in the SLC4A11 gene may result in the disease.[7][13][14] However, not all patients with CHED have been found to carry mutations of SLC4A11, which suggests genetic heterogeneity.[6][7][13] For example, one study of a mutation screening in 21 Iranian patients diagnosed with CHED, noted that in one patient, there was no SLC4A11 mutation. Instead, an MPDZ mutation was identified, which was similarly found in her mother, who had Fuchs Dystrophy.[13] Another study reported that screening of the SLC4A11 promoter region in 20 families affected with CHED resulted in no pathogenic SLC4A11 variants.[6]
In 2016, a novel variant of CHED was reported in a 45-year-old woman that resulted in delayed onset of the disease. She reported poor bilateral vision since childhood and rapid decline of vision the past 5-years. CHED was confirmed by slit-lamp examination, histopathology, genetic screening, and clinical correlation. Genetic testing confirmed new compound heterozygous mutations affecting the SLC4A11 gene.[15] Due to this single and unusual presentation, it is difficult to interpret the reason for a delayed onset. It has been suggested that the woman’s lack of access to care and socioeconomic status contributed to her delay in seeking care, but it has also been considered that compound heterozygous mutations of SLC4A11 may only mildly affect BTR function, resulting in delayed onset of CHED signs and symptoms.[15]
Risk Factors
Family history positive for CHED and consanguineous parents increases a neonates’ risk of being born with the disease. It is recommended that families with children diagnosed with CHED be offered genetic counseling due to a 25% risk of recurring in future offspring.[3]
Epidemiology
An analysis published in 1998 analyzed 1,124,654 births in Spain and reported the incidence of congenital corneal opacities to be 3.11/100,000 newborns.[16] The current prevalence and incidence of CHED are unknown. Cases that have been identified are primarily among children with family history for corneal dystrophy and consanguineous parents from Saudi Arabia, Pakistan, India, and Ireland.[3][17][18][19]
Pathophysiology
Congenital hereditary endothelial dystrophy manifests in the neonatal period as corneal edema and opacification. Edema can result in 2 to 3 times the normal corneal thickness.[1] Corneal endothelium prevents stromal edema by countering osmotically driven fluid from the aqueous humor into the stromal collagen. The SLC4A11 gene is active in corneal endothelium, and codes for bicarbonate transporter-related protein-1 (BTR1) that acts as a sodium-borate cotransporter to relieve excess water from the stroma. In CHED, BTR-1 is dysfunctional and often fails to reach the plasma membrane to perform its purpose.[6][20] Histopathologically, corneal opacification due to diffuse epithelial and stromal edema and thickening of Descemet's membrane due to abnormal endothelial secretion are noted. Sparse, fibrotic endothelial cells are also observed.[1][3]
History and Physical
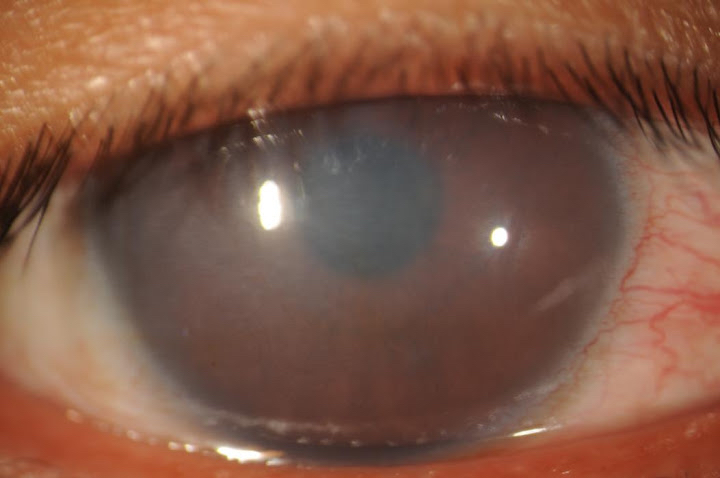
For a young patient with corneal opacification, the onset of symptoms and family history should be elicited from the patient’s family. In older untreated children, it is essential to discuss the timing of onset, as well as the progression of symptoms of reduced vision, amblyopia, nystagmus, and possible hearing loss.[1][3][5] Pendular nystagmus may be seen early in infancy but is not obvious and difficult to discern due to the small amplitude waveform.[21][22] It has been observed that nystagmus is more often found among patients with severe corneal clouding.[22] On slit-lamp examination, congenital hereditary endothelial dystrophy manifests as bilateral corneal edema, presenting in the neonatal period. The cornea may show a blueish-gray ground-glass appearance but can be as severe as complete corneal opacification.[23] (Figure 1)
Evaluation
Endothelial function and permeability can be further assessed by the fluoro-photometrical method.[3][24] Intraocular pressure should be evaluated due to the possibility of glaucoma, but the provider should be aware of artifactually elevated pressure due to thickened corneas.[23][25] In such cases, supporting markers of congenital glaucoma such as increased corneal diameter, Haab striae, and buphthalmos should be assessed.[23]
Audiometric monitoring should also be considered due to the possibility of Harboyan syndrome, a form of congenital hereditary endothelial dystrophy associated with sensorineural hearing loss. Hearing deficit is not usually found until 10 to 25 years old, and there have been no reported cases of prelingual deafness.[1][3][12] Some studies suggest hearing loss will develop in all individuals with CHED over time, as has been revealed in mice with disrupted SLC4A11.[1][12]
Treatment / Management
The definitive treatment for congenital hereditary endothelial dystrophy is currently surgical, either by penetrating keratoplasty (PK) or endothelial keratoplasty (EK). PK is a full-thickness graft and has traditionally been the mainstay of treatment.[26] EK encompasses Descemet stripping automated endothelial keratoplasty (DSAEK) and Descemet membrane endothelial keratoplasty (DMEK). DSAEK grafting, where only the endothelium and part of the posterior stroma are transplanted, has shown to be a viable option for patients with CHED with clear advantages over PK.[2][27] DMEK is a newer style of EK surgery that has advantages over DSAEK, but only a few studies have been reported regarding its use for pediatric treatment.[28][29](B2)
PK is technically challenging in the pediatric population, both operatively and postoperatively.[2][26][30] Children have low scleral rigidity and positive vitreous pressure, which are associated with frequent suture loosening and increased risk for infection.[2] Children also have increased risk for rejection and dehiscence, and are more likely to have nonadherence to postoperative directions.[26][30] Unpredictable astigmatism post-surgery is a common problem, even with clear donor grafts.[2] Older children tend to have a better prognosis, leading some physicians electing to delay surgery as long as possible, which consequently increases the risk of amblyopia and poor visual outcomes despite intervention.[2][26][31] In 2018, a retrospective review reported poor outcomes with PK among 14 patients from Ireland with CHED from 1978 through 2013. The study demonstrated a high-risk of long-term graft failure within 37-months, with graft survival rates of 37.5% for primary graft and 33% for regrafting.[18] Of the 24 eyes evaluated, postoperative best-corrected visual acuity (BCVA) was the same or worse than preoperative BCVA in 79% of eyes treated with PK.[18](B2)
DSAEK grafting, where only the endothelium and part of the posterior stroma are transplanted, has proven to be a viable alternative to PK in CHED. Studies suggest improvement in clinical outcomes without significant complications.[2][4][24] This procedure involves stripping of the host’s endothelium and replacing it with a donor endothelium graft.[2] This procedure is becoming preferred to PK due to its advantages of more rapid visual recovery, more stable refractive error, decreased likelihood of amblyopia and decreased traumatic globe rupture due to the small incision.[2][4][24][27] Ashar et al. reported a distinct advantage of reduced postoperative astigmatism in CHED DSAEK patients, compared to PK.[27] In a study by Yang et al., infants less than one year of age showed decreased complications, faster recovery, and better outcomes of DSAEK compared to children more than one year of age. It is suggested that the difference may be that infants are more likely to remain supine for graft survival as well as begin amblyopia treatment and rehabilitation at an earlier age.[2] The average BCVA prior to DSAEK was logMAR 1.03 (greater than 20/200), with improved BCVA postoperatively reported as logMAR 0.54 (20/63) for children and logMAR 0.32 (20/40) for infants. Age-specific timing recommendations for DSAEK remains debatable, but earlier intervention is recommended for better visual rehabilitation. Factors such as patient age, disease severity, and surgeon ability should be taken into consideration.[2](B2)
However, DSAEK is not without complications. Children have a clear crystallin lens, a smaller anterior chamber, and increased difficulty to identify and strip the Descemet’s membrane, especially in infants.[2][4] Some surgeons modify the DSAEK procedure for patients with CHED and elect not to strip the Descemet’s membrane of the infant host, and instead directly attach the donor graft to the posterior surface of the cornea.[32] This method is associated with increased postoperative graft dislocation, but overall good long-term outcomes.[32] Acute postoperative complications for DSAEK include lenticule/graft detachment, which is managed with re-bubbling but may ultimately require PK due to stromal scarring if detachment continues.[2][33] For young children, a case series suggests extra steps to ensure intraoperative adhesion, such as a 15-minute corneal massage, large anterior chamber air bubble, possible venting incision, and additional days with postoperative eyeshield.[34](B2)
Descemet’s membrane endothelial keratoplasty (DMEK) is an alternative EK procedure. The graft is prepared by manually stripping the Descemet’s membrane with fine tweezers from the donor cornea.[35] The graft is extremely fragile and thin, and at only 15-um thick, it rolls up and can be inserted via a smaller incision than used in DSAEK. Compared to DSAEK, the DMEK graft has no stromal layer, which avoids incompatible convergence of the host and donor stromal fibers.[36] DMEK has shown to provide better functional results than both DSAEK and PK, as well as decreased long-term graft rejection.[37] However, the procedure is surgically demanding and more challenging to manage among infants. A 2017 case report documented an unsuccessful DMEK in a 4-month old infant with PPCD due to graft dissociation five days postoperatively.[28] Rebubbling of the graft was unsuccessful, and the infant was later treated with a DSAEK graft.[28] A case report in 2014 documented successful DMEK in a 12-year old patient with no postoperative complications by 6-months.[29] DMEK treatment, specifically for patients with CHED, remains an area of high interest.(B2)
Some exciting studies have suggested that SLC4A11 mutations that result in impaired-folding of the BTR-1 protein being retained in the endoplasmic reticulum (ER) can be corrected nonsurgically with non-steroidal anti-inflammatory drugs (NSAIDS).[11][20][38] In a 2015 study, Glafenine was shown to significantly correct folding defects and rescue SLC4A11 cell surface functional activity.[11][38] Another study in 2018 tested five NSAIDS for their ability to correct ER-retained mutants and found diclofenac and nepafenac to be highly effective at restoring water-flux activity similar to wild-type SLC4A11.[20] The limitation of this potential benefit depends on whether an individual’s SLC4A11 mutation results in misfolded proteins that are retained in the endoplasmic reticulum. This is a promising treatment since the majority of CHED cases are associated with retained-ER proteins.[11] CHED is disproportionately found in developing countries where consanguineous marriages are common, and diclofenac therapy would be relatively accessible and cheap.[20] Further clinical trials are necessary to establish appropriate dosing regimens.(B3)
Differential Diagnosis
The differential diagnosis for pediatric corneal opacities is broad, and a classification system can aid the organization of the diseases. One method is via the mnemonic STUMPED: Sclerocornea, Tears in Descemet's membrane, Ulcers, Metabolic, Peters anomaly, Endothelial dystrophy, Dermoid.[39] Another approach is to classify corneal opacities by primary congenital and secondary congenital and acquired.[40]
Sclerocornea
A congenital disorder resulting in poor development of the anterior chamber presents with peripheral corneal opacity resembling sclera. The central cornea is often spared.[41] Identifying characteristics include a flattened cornea with peripheral scleralization resulting in the loss of an identifiable limbus.[40][41]
Tears in Descemet's Membrane
This most commonly is caused by birth trauma associated with forceps assisted delivery and presents with clouding and edema due to the rupture of Descemet's membrane. Linear and vertical Haab striae (breaks in the Descemet's membrane) may be identified.[40] Periorbital soft-tissue damage will also be present in many cases.[41]
Congenital glaucoma can present at birth as corneal clouding with elevated IOP. It is essential to identify markers for congenital glaucoma such as buphthalmos, optic disk cupping, curvilinear Haab's striae oriented horizontally, and concentric to the limbus, and enlarged corneal diameter.[23][40] Thickened corneas due to endothelial dystrophy can result in a falsely elevated IOP per tonometry, and an incorrect diagnosis of glaucoma may be given.[21] Before a diagnosis of glaucoma can be given to a child with corneal opacification, primary corneal disease must first be ruled out.[21]
Ulcers
The presence of corneal ulceration should increase suspicion for an infectious cause. Common infections include herpes simplex keratitis and bacterial keratitis.[40][41] Herpes keratitis presents within the first 2-weeks of birth and is characterized as a cloudy cornea with a large epithelial ulcer.[40] Bacterial keratitis can result in scarring of the cornea and may require surgery.[40]
Metabolic
Mucopolysaccharidoses, cystinosis, and mucolipidosis IV should be considered. Mucopolysaccharidosis is a lysosomal storage disorder that can result in corneal clouding within the first year of life due to the accumulation of glycosaminoglycans.[42] Cystinosis is characterized by elevated cystine levels and systemic deposition. Iridescent elongated corneal crystals can accumulate in the cornea by 1-year of age.[43] Mucolipidosis IV presents early after birth and presents with severe psychomotor delay requiring hospitalization.[40]
Peters Anomaly
This is identified by centralized vascular corneal opacity due to the failure of the cornea to separate from the iris and lens.[40][44] The patient may also present with iridocorneal adhesions, cataracts, and corneolenticular adhesions.[40] Peters plus syndrome is more severe and associated with cleft palate, short stature, abnormal ears, and mental retardation.[44]
Endothelial Dystrophy
CHED is distinguished from other corneal opacities as diffuse, nonbullous edema with corneal opacification ranging from blue-gray to complete opacity. Opacification remains consistent over time. Due to thickened corneas, intraocular pressure measurements may be artifactually elevated.[40] In 2016, a case-report presented a 10-year old boy with pendular nystagmus and iris colobomas being treated for glaucoma, but only had CHED. This case highlights the importance of careful observation and a thorough differential workup.[21]
Posterior polymorphous corneal dystrophy (PPCD) is autosomal dominant and widely variable in presentation.[6] It generally presents with blister-like lesions of the corneal endothelium and edema in the first few years of life. Histology reveals proliferating epithelialization of endothelial cells into the trabecular meshwork that may cause secondary glaucoma.[3][45] Unlike CHED, PPCD is described as progressive corneal clouding that develops within the first years of life, rarely at birth. Clarifying parental history or positive findings of PPCD in an affected parent may help make the diagnosis.[40]
Congenital hereditary stromal dystrophy (CHSD) in an autosomal dominant disorder that presents as diffuse corneal opacity with a central flaky appearance in the stroma and is nonprogressive from birth.[40][41]
Dermoid
Dermoids are choriostomas that present near the limbus and may obstruct the visual axis. Size can vary, but large dermoids often affect vision and require surgery to prevent amblyopia.[40][41]
Prognosis
Prognosis is determined by a multitude of factors, including disease severity, age at treatment, and existing comorbidities. Surgery performed at an earlier age avoids amblyopia and allows for earlier visual rehabilitation for improved visual development, but is associated with increased risk for surgical complications.[2][32] It is widely accepted that children with congenital hereditary endothelial dystrophy who undergo PK treatment have excellent graft survival and are significantly more likely to achieve ambulatory vision compared to non-CHED PK treatment.[26] However, this claim is countered by two 2017 studies reporting very poor outcomes of PK in patients with CHED due to graft rejection and poor BCVA postoperatively.[18][31] Despite that PK has been the traditional treatment, DSAEK procedures are becoming more preferred due to better prognosis, decreased complications, faster corneal recovery, and better corrected visual acuity.[2][27] The probability of rejection for corneal grafts at 1 and 2 years postoperatively has been reported as 1% and 1% for DMEK, 8% and 12% for DSAEK, and 14% and 18% for PK.[37]
Complications
Untreated congenital hereditary endothelial dystrophy can result in poor visual development and occlusive amblyopia. Although the risk of developing amblyopia decreases the younger a child is treated, earlier interventions are associated with increased risk of graft dehiscence, suture induced amblyopia, and increased exposure to anesthesia.[27][46]
Astigmatism is a significant risk with corneal grafting. The risk for astigmatism is highest in PK procedures and can sometimes be severe enough to result in amblyopia.[27] DSAEK has been associated with a decreased risk of developing astigmatism.[2][27] Elevated intraocular pressure requiring medication and a postoperative hyperopic shift has been reported in patients with CHED receiving DSAEK.[47]
Lens trauma during EK grafting is possible due to a child’s clear, crystalline lens. Trauma can result in the development of an iatrogenic cataract. One method used to protect the lens during EK is with pilocarpine, a cholinergic agonist, which induces miosis.[46]
Deterrence and Patient Education
Due to the genetic inheritance of CHED, it is recommended that patients with a positive family history be offered genetic counseling.[3]
Pearls and Other Issues
- Congenital hereditary endothelial dystrophy is a rare, autosomal recessive disorder causing endothelial dysfunction resulting in corneal opacification.
- Pathognomonic findings include bilateral, symmetric corneal opacification presenting in the immediate neonatal period with increased corneal thickness, corneal edema, and thickening of the Descemet’s membrane.
- Current treatment requires grafting, either by PK or EK. DSAEK is associated with decreased postoperative complications and faster corneal recovery.
- New studies have shown certain Nonsteroidal Anti-inflammatories such as glafenine, diclofenac, and nepafenac, to be highly effective at restoring water-flux activity in certain mutational types of patients with CHED and are promising potential treatments
- Early treatment avoids amblyopia but may increase surgical risk and postoperative complications.
Enhancing Healthcare Team Outcomes
Congenital hereditary endothelial dystrophy is a rare disease and primarily found in developing countries among already affected families with consanguinity.[3] Family education and genetic counseling among affected populations are essential for the prevention of CHED, and require an interprofessional team to identify, treat, and educate. It is important to identify CHED early to prevent permanent disability due to poor visual development. Primary care providers who identify corneal opacification or note nystagmus in an infant or child should consult ophthalmologists for evaluation. Communication among the healthcare team, pediatricians, ophthalmologists, geneticists, and medical scientists will help identify CHED in affected families, provide prompt treatment, and prevent future occurrence.
Media
(Click Image to Enlarge)
Congenital Hereditary Endothelial Dystrophy Contributed by Majid Moshirfar, MD
References
Patel SP, Parker MD. SLC4A11 and the Pathophysiology of Congenital Hereditary Endothelial Dystrophy. BioMed research international. 2015:2015():475392. doi: 10.1155/2015/475392. Epub 2015 Sep 16 [PubMed PMID: 26451371]
Yang F, Hong J, Xiao G, Feng Y, Peng R, Wang M, Qu H. Descemet Stripping Endothelial Keratoplasty in Pediatric Patients with Congenital Hereditary Endothelial Dystrophy. American journal of ophthalmology. 2020 Jan:209():132-140. doi: 10.1016/j.ajo.2019.08.010. Epub 2019 Aug 26 [PubMed PMID: 31465754]
Desir J, Abramowicz M. Congenital hereditary endothelial dystrophy with progressive sensorineural deafness (Harboyan syndrome). Orphanet journal of rare diseases. 2008 Oct 15:3():28. doi: 10.1186/1750-1172-3-28. Epub 2008 Oct 15 [PubMed PMID: 18922146]
Ashar JN, Ramappa M, Vaddavalli PK. Paired-eye comparison of Descemet's stripping endothelial keratoplasty and penetrating keratoplasty in children with congenital hereditary endothelial dystrophy. The British journal of ophthalmology. 2013 Oct:97(10):1247-9. doi: 10.1136/bjophthalmol-2012-302602. Epub 2013 Apr 23 [PubMed PMID: 23613513]
Level 2 (mid-level) evidenceSchmedt T, Silva MM, Ziaei A, Jurkunas U. Molecular bases of corneal endothelial dystrophies. Experimental eye research. 2012 Feb:95(1):24-34. doi: 10.1016/j.exer.2011.08.002. Epub 2011 Aug 10 [PubMed PMID: 21855542]
Level 3 (low-level) evidenceAldave AJ, Han J, Frausto RF. Genetics of the corneal endothelial dystrophies: an evidence-based review. Clinical genetics. 2013 Aug:84(2):109-19. doi: 10.1111/cge.12191. Epub 2013 Jun 10 [PubMed PMID: 23662738]
Paliwal P, Sharma A, Tandon R, Sharma N, Titiyal JS, Sen S, Nag TC, Vajpayee RB. Congenital hereditary endothelial dystrophy - mutation analysis of SLC4A11 and genotype-phenotype correlation in a North Indian patient cohort. Molecular vision. 2010 Dec 31:16():2955-63 [PubMed PMID: 21203343]
Level 3 (low-level) evidenceLopez IA, Rosenblatt MI, Kim C, Galbraith GC, Jones SM, Kao L, Newman D, Liu W, Yeh S, Pushkin A, Abuladze N, Kurtz I. Slc4a11 gene disruption in mice: cellular targets of sensorineuronal abnormalities. The Journal of biological chemistry. 2009 Sep 25:284(39):26882-96. doi: 10.1074/jbc.M109.008102. Epub 2009 Jul 8 [PubMed PMID: 19586905]
Level 3 (low-level) evidenceJalimarada SS,Ogando DG,Vithana EN,Bonanno JA, Ion transport function of SLC4A11 in corneal endothelium. Investigative ophthalmology [PubMed PMID: 23745003]
Level 3 (low-level) evidenceChaurasia S, Ramappa M, Annapurna M, Kannabiran C. Coexistence of Congenital Hereditary Endothelial Dystrophy and Fuchs Endothelial Corneal Dystrophy Associated With SLC4A11 Mutations in Affected Families. Cornea. 2020 Mar:39(3):354-357. doi: 10.1097/ICO.0000000000002183. Epub [PubMed PMID: 31714402]
Alka K, Casey JR. Molecular phenotype of SLC4A11 missense mutants: Setting the stage for personalized medicine in corneal dystrophies. Human mutation. 2018 May:39(5):676-690. doi: 10.1002/humu.23401. Epub 2018 Feb 2 [PubMed PMID: 29327391]
Level 3 (low-level) evidenceSiddiqui S, Zenteno JC, Rice A, Chacón-Camacho O, Naylor SG, Rivera-de la Parra D, Spokes DM, James N, Toomes C, Inglehearn CF, Ali M. Congenital hereditary endothelial dystrophy caused by SLC4A11 mutations progresses to Harboyan syndrome. Cornea. 2014 Mar:33(3):247-51. doi: 10.1097/ICO.0000000000000041. Epub [PubMed PMID: 24351571]
Moazzeni H,Javadi MA,Asgari D,Khani M,Emami M,Moghadam A,Panahi-Bazaz MR,Hosseini Tehrani M,Karimian F,Hosseini B,Nekuie Moghadam T,Hassanpour H,Akbari MT,Elahi E, Observation of nine previously reported and 10 non-reported {i}SLC4A11{/i} mutations among 20 Iranian CHED probands and identification of an {i}MPDZ{/i} mutation as possible cause of CHED and FECD in one family. The British journal of ophthalmology. 2019 Aug 16; [PubMed PMID: 31420327]
Sultana A, Garg P, Ramamurthy B, Vemuganti GK, Kannabiran C. Mutational spectrum of the SLC4A11 gene in autosomal recessive congenital hereditary endothelial dystrophy. Molecular vision. 2007 Jul 26:13():1327-32 [PubMed PMID: 17679935]
Kumawat BL, Gupta R, Sharma A, Sen S, Gupta S, Tandon R. Delayed onset of congenital hereditary endothelial dystrophy due to compound heterozygous SLC4A11 mutations. Indian journal of ophthalmology. 2016 Jul:64(7):492-5. doi: 10.4103/0301-4738.190100. Epub [PubMed PMID: 27609159]
Bermejo E, Martínez-Frías ML. Congenital eye malformations: clinical-epidemiological analysis of 1,124,654 consecutive births in Spain. American journal of medical genetics. 1998 Feb 17:75(5):497-504 [PubMed PMID: 9489793]
Level 2 (mid-level) evidenceHemadevi B,Veitia RA,Srinivasan M,Arunkumar J,Prajna NV,Lesaffre C,Sundaresan P, Identification of mutations in the SLC4A11 gene in patients with recessive congenital hereditary endothelial dystrophy. Archives of ophthalmology (Chicago, Ill. : 1960). 2008 May; [PubMed PMID: 18474783]
AlArrayedh H, Collum L, Murphy CC. Outcomes of penetrating keratoplasty in congenital hereditary endothelial dystrophy. The British journal of ophthalmology. 2018 Jan:102(1):19-25. doi: 10.1136/bjophthalmol-2016-309565. Epub 2017 May 6 [PubMed PMID: 28478395]
Mohamed MD, McKibbin M, Jafri H, Rasheed Y, Woods CG, Inglehearn CF. A new pedigree with recessive mapping to CHED2 locus on 20p13. The British journal of ophthalmology. 2001 Jun:85(6):758-9 [PubMed PMID: 11439918]
Level 3 (low-level) evidenceAlka K, Casey JR. Ophthalmic Nonsteroidal Anti-Inflammatory Drugs as a Therapy for Corneal Dystrophies Caused by SLC4A11 Mutation. Investigative ophthalmology & visual science. 2018 Aug 1:59(10):4258-4267. doi: 10.1167/iovs.18-24301. Epub [PubMed PMID: 30140924]
Level 3 (low-level) evidenceKhan AO,Aldahmesh MA,Alkuraya F, Congenital hereditary endothelial dystrophy, not glaucoma, in a child with iris colobomas. Journal of AAPOS : the official publication of the American Association for Pediatric Ophthalmology and Strabismus. 2016 Aug; [PubMed PMID: 27373217]
Judisch GF, Maumenee IH. Clinical differentiation of recessive congenital hereditary endothelial dystrophy and dominant hereditary endothelial dystrophy. American journal of ophthalmology. 1978 May:85(5 Pt 1):606-12 [PubMed PMID: 306759]
Level 3 (low-level) evidenceNischal KK. Genetics of Congenital Corneal Opacification--Impact on Diagnosis and Treatment. Cornea. 2015 Oct:34 Suppl 10():S24-34. doi: 10.1097/ICO.0000000000000552. Epub [PubMed PMID: 26352876]
Ehlers N, Módis L, Møller-Pedersen T. A morphological and functional study of Congenital Hereditary Endothelial Dystrophy. Acta ophthalmologica Scandinavica. 1998 Jun:76(3):314-8 [PubMed PMID: 9686844]
Khan AO,Al-Shehah A,Ghadhfan FE, High measured intraocular pressure in children with recessive congenital hereditary endothelial dystrophy. Journal of pediatric ophthalmology and strabismus. 2010 Jan-Feb; [PubMed PMID: 20128551]
Level 3 (low-level) evidenceAl-Ghamdi A, Al-Rajhi A, Wagoner MD. Primary pediatric keratoplasty: indications, graft survival, and visual outcome. Journal of AAPOS : the official publication of the American Association for Pediatric Ophthalmology and Strabismus. 2007 Feb:11(1):41-7 [PubMed PMID: 17307682]
Level 2 (mid-level) evidenceAshar JN, Madhavi Latha K, Vaddavalli PK. Descemet's stripping endothelial keratoplasty (DSEK) for children with congenital hereditary endothelial dystrophy: surgical challenges and 1-year outcomes. Graefe's archive for clinical and experimental ophthalmology = Albrecht von Graefes Archiv fur klinische und experimentelle Ophthalmologie. 2012 Sep:250(9):1341-5. doi: 10.1007/s00417-012-2014-8. Epub 2012 Apr 19 [PubMed PMID: 22527319]
Level 2 (mid-level) evidenceHermina Strungaru M, Ali A, Rootman D, Mireskandari K. Endothelial keratoplasty for posterior polymorphous corneal dystrophy in a 4-month-old infant. American journal of ophthalmology case reports. 2017 Sep:7():23-26. doi: 10.1016/j.ajoc.2017.05.001. Epub 2017 May 4 [PubMed PMID: 29260073]
Level 3 (low-level) evidenceGonnermann J, Klamann MK, Maier AK, Bertelmann E, Schroeter J, von Au K, Joussen AM, Torun N. Descemet membrane endothelial keratoplasty in a child with corneal endothelial dysfunction in Kearns-Sayre syndrome. Cornea. 2014 Nov:33(11):1232-4. doi: 10.1097/ICO.0000000000000252. Epub [PubMed PMID: 25211357]
Level 3 (low-level) evidenceHuang PT. Penetrating keratoplasty in infants and children. Journal of AAPOS : the official publication of the American Association for Pediatric Ophthalmology and Strabismus. 2007 Feb:11(1):5-6 [PubMed PMID: 17307675]
Gulias-Cañizo R, Gonzalez-Salinas R, Hernandez-Zimbron LF, Hernandez-Quintela E, Sanchez-Huerta V. Indications and outcomes of pediatric keratoplasty in a tertiary eye care center: A retrospective review. Medicine. 2017 Nov:96(45):e8587. doi: 10.1097/MD.0000000000008587. Epub [PubMed PMID: 29137083]
Level 2 (mid-level) evidenceBusin M, Beltz J, Scorcia V. Descemet-stripping automated endothelial keratoplasty for congenital hereditary endothelial dystrophy. Archives of ophthalmology (Chicago, Ill. : 1960). 2011 Sep:129(9):1140-6. doi: 10.1001/archophthalmol.2011.114. Epub 2011 May 9 [PubMed PMID: 21555597]
Kaur M, Titiyal JS, Gagrani M, Shaikh F, Agarwal T, Sinha R, Sharma N. Repeat keratoplasty in failed Descemet stripping automated endothelial keratoplasty. Indian journal of ophthalmology. 2019 Oct:67(10):1586-1592. doi: 10.4103/ijo.IJO_1729_18. Epub [PubMed PMID: 31546486]
Mittal V, Mittal R. Challenges in pediatric endothelial keratoplasty. Indian journal of ophthalmology. 2014 Feb:62(2):251-4. doi: 10.4103/0301-4738.128638. Epub [PubMed PMID: 24618494]
Level 3 (low-level) evidenceMaier P, Reinhard T, Cursiefen C. Descemet stripping endothelial keratoplasty--rapid recovery of visual acuity. Deutsches Arzteblatt international. 2013 May:110(21):365-71. doi: 10.3238/arztebl.2013.0365. Epub 2013 May 24 [PubMed PMID: 23795211]
Melles GR, Ong TS, Ververs B, van der Wees J. Descemet membrane endothelial keratoplasty (DMEK). Cornea. 2006 Sep:25(8):987-90 [PubMed PMID: 17102683]
Level 3 (low-level) evidenceAnshu A, Price MO, Price FW Jr. Risk of corneal transplant rejection significantly reduced with Descemet's membrane endothelial keratoplasty. Ophthalmology. 2012 Mar:119(3):536-40. doi: 10.1016/j.ophtha.2011.09.019. Epub 2012 Jan 3 [PubMed PMID: 22218143]
Level 2 (mid-level) evidenceChiu AM, Mandziuk JJ, Loganathan SK, Alka K, Casey JR. High Throughput Assay Identifies Glafenine as a Corrector for the Folding Defect in Corneal Dystrophy-Causing Mutants of SLC4A11. Investigative ophthalmology & visual science. 2015 Dec:56(13):7739-53. doi: 10.1167/iovs.15-17802. Epub [PubMed PMID: 26641551]
Level 3 (low-level) evidenceMiao S, Lin Q, Liu Y, Song YW, Zhang YN, Pan ZQ. Clinicopathologic Features and Treatment Characteristics of Congenital Corneal Opacity Infants and Children Aged 3 Years or Less: A Retrospective Single Institution Analysis. Medical principles and practice : international journal of the Kuwait University, Health Science Centre. 2020:29(1):18-24. doi: 10.1159/000501763. Epub 2019 Jun 28 [PubMed PMID: 31247621]
Level 2 (mid-level) evidenceNischal KK. A new approach to the classification of neonatal corneal opacities. Current opinion in ophthalmology. 2012 Sep:23(5):344-54. doi: 10.1097/ICU.0b013e328356893d. Epub [PubMed PMID: 22871880]
Level 3 (low-level) evidenceKurji K, Damji K. Ophthaproblem. Can you identify this condition? Primary congenital glaucoma. Canadian family physician Medecin de famille canadien. 2012 Apr:58(4):409, 412-3 [PubMed PMID: 22499817]
Level 3 (low-level) evidenceTomatsu S, Pitz S, Hampel U. Ophthalmological Findings in Mucopolysaccharidoses. Journal of clinical medicine. 2019 Sep 14:8(9):. doi: 10.3390/jcm8091467. Epub 2019 Sep 14 [PubMed PMID: 31540112]
Bose S, Yeo DCM, Wijetilleka S. Using two smartphones to look for corneal cystine crystals. Digital journal of ophthalmology : DJO. 2019 Jan:25(1):12-15. doi: 10.5693/djo.02.2019.02.003. Epub 2019 Mar 29 [PubMed PMID: 31080371]
Bhandari R, Ferri S, Whittaker B, Liu M, Lazzaro DR. Peters anomaly: review of the literature. Cornea. 2011 Aug:30(8):939-44. doi: 10.1097/ICO.0b013e31820156a9. Epub [PubMed PMID: 21448066]
Level 3 (low-level) evidenceDavidson AE, Liskova P, Evans CJ, Dudakova L, Nosková L, Pontikos N, Hartmannová H, Hodaňová K, Stránecký V, Kozmík Z, Levis HJ, Idigo N, Sasai N, Maher GJ, Bellingham J, Veli N, Ebenezer ND, Cheetham ME, Daniels JT, Thaung CM, Jirsova K, Plagnol V, Filipec M, Kmoch S, Tuft SJ, Hardcastle AJ. Autosomal-Dominant Corneal Endothelial Dystrophies CHED1 and PPCD1 Are Allelic Disorders Caused by Non-coding Mutations in the Promoter of OVOL2. American journal of human genetics. 2016 Jan 7:98(1):75-89. doi: 10.1016/j.ajhg.2015.11.018. Epub 2015 Dec 31 [PubMed PMID: 26749309]
Panahi-Bazaz M, Sharifipour F, Malekahmadi M. Modified Descemet's Stripping Automated Endothelial Keratoplasty for Congenital Hereditary Endothelial Dystrophy. Journal of ophthalmic & vision research. 2014 Oct-Dec:9(4):522-5. doi: 10.4103/2008-322X.150836. Epub [PubMed PMID: 25709783]
Mohebbi M, Nabavi A, Fadakar K, Hashemi H. Outcomes of Descemet-Stripping Automated Endothelial Keratoplasty in Congenital Hereditary Endothelial Dystrophy. Eye & contact lens. 2020 Jan:46(1):57-62. doi: 10.1097/ICL.0000000000000604. Epub [PubMed PMID: 31008826]