Introduction
Vitamin-D, calcium, and phosphorus are the main factors that influence bone maturation and mineralization. Defective mineralization can lead to rickets and/or osteomalacia. Rickets is characterized by a defect in mineralization and the widening of the epiphyseal plates. Osteomalacia, however, is a defect in the mineralization of the bone matrix. Both rickets and osteomalacia usually occur together in children. Rickets occurs exclusively in children, whereas adults develop osteomalacia after the epiphyseal plate fusion.[1] Whistler, Boate, Glisson, and their colleagues, Fellows of the Royal College of Physicians, London, in the seventeenth century, were the first to describe rickets in the medical literature.[2] Nutritional rickets is the most common cause of bone disease all over the world.[3]
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
Vitamin D deficiency is, by far, the most common cause of nutritional rickets.[4] Rarely, nutritional deficiency of calcium or phosphorus can result in rickets. Other less frequent causes of rickets include genetic causes, drug-induced rickets, and rickets secondary to liver diseases. Medications that impair vitamin D metabolisms such as diphenylhydantoin and rifampicin can result in rickets.[5]
Based on the biochemical profile, rickets can be classified into calcipenic, phosphopenic, and rickets due to inhibited mineralization.[6]
- Deficiency of calcium results in calcipenic rickets, and vitamin D deficiency is the most common etiology for calcipenic rickets. Calcipenic rickets may result from inadequate dietary calcium intake, which is reported in a few developing countries.[6] Calcipenic rickets may also result from poor calcium absorption, such as in children with malabsorption syndromes, especially celiac disease and cystic fibrosis.[2] Rickets can be the first presenting manifestation in patients with celiac disease.[7] Additionally, calcipenic rickets may result from a genetic defect of vitamin D metabolism, either from a failure of vitamin D to switch to its active form (1,25-dihydroxy vitamin D) or due to end-organ resistance. Low serum calcium is a common phenomenon in calcipenic rickets that stimulates the secretion of parathyroid hormone (PTH), which may result in the normalization of serum calcium. In the long term, this secondary hyperparathyroidism results in the internalization of sodium-dependent phosphate co-transporter proteins in the renal tubules. Subsequently, it causes renal phosphate loss and hypophosphatemia.[8]
- Phosphopenic rickets, on the other hand, is caused by conditions that cause chronic low serum phosphate levels, either from impaired intestinal absorption or, more commonly, from increased renal loss. Phosphate is abundant in our regular food, so phosphate deficiency does not usually occur from dietary insufficiency in healthy individuals. In premature children, dietary phosphate deficiency can result in osteopenia of prematurity. Low serum phosphate levels occur in (1) conditions that increase the production or decrease degradation of fibroblast growth factor 23 (FGF23), a hormone that reduces the reabsorption and increases the excretion of phosphate in the renal tubules. (2) increased urinary phosphate loss due to mutations that cause inactivation of sodium-dependent phosphate transporters in the kidneys. Both conditions increase the urinary phosphate loss resulting in chronic hypophosphatemia. Both calcipenic rickets and phosphopenic rickets are characterized by hypophosphatemia, which eventually causes the clinical and radiological bone changes characteristic of rickets (rachitic changes).[8] (more details in pathophysiology section)
- Inhibited mineralization rickets occurs when there is a defect in growth plate mineralization in the presence of normal calcium and phosphate concentrations. It may result from several predisposing factors including, hereditary hypophosphatasia, medications (first-generation bisphosphonates), and toxicities from aluminum and fluoride.[6]
Genetic causes of rickets may be classified broadly into two types: vitamin D-dependent rickets (calcipenic type), and congenital hypophosphatemic rickets (phosphopenic type).[9][10]
Vitamin D-dependent rickets:[10] This group is characterized by defects in either synthesis of the active form of vitamin D (1,25-dihydroxy vitamin D), or defect in vitamin D receptor (VDR), or vitamin D-VDR interactions.
- Vitamin D-dependent rickets type I A (VDDR1A): This type is previously known as hereditary pseudo-vitamin D deficiency. It is inherited by an autosomal recessive mutation in the CYP27B1. In this form of rickets, 25-hydroxyvitamin D cannot be converted to 1,25-dihydroxy vitamin D due to a deficiency in the 1-alpha hydroxylase enzyme, which is required for vitamin D biosynthesis in the kidney.
- Vitamin D-dependent rickets type I B (VDDR1B): This type is also referred to as vitamin D hydroxylation-deficient rickets. It is a rare condition inherited by an autosomal recessive mutation in the CYP2R1 gene. In this form of rickets, vitamin D cannot be converted into 25-hydroxyvitamin D and later to 1,25-dihydroxy vitamin D, due to a deficiency in the 25-hydroxylase enzyme, which is another enzyme that required for vitamin D biosynthesis present in the liver.
- Vitamin D-dependent rickets type II A (VDDR2A): This type of rickets is also known as hereditary vitamin D-resistant rickets. It is inherited as an autosomal recessive condition. It is characterized by end-organ resistance to the active form of vitamin D. VDDR2A is caused by VDR gene mutations, and these mutations alter the VDR, often preventing the receptor from interacting with 1,25-dihydroxy vitamin D. As a result, VDR cannot regulate gene activity, even with normal amounts of 1,25-dihydroxy vitamin D in the body.
- Vitamin D-dependent rickets type II B (VDDR2B): This is an unusual form of vitamin D-dependent rickets. It is caused by overexpression of a nuclear protein that interferes with normal vitamin D function, and the exact pathogenesis is unclear.
Congenital hypophosphatemic rickets:[10] In this type, the defect in bone mineralization is caused by hypophosphatemia (secondary to renal phosphate loss). It is classified into two groups, FGF-23-dependent hypophosphatemic rickets, and FGF-23-independent hypophosphatemic rickets.
- FGF-23-dependent hypophosphatemic rickets: It is characterized by an abnormality in phosphatonin, which regulates phosphate homeostasis. It is further classified into three forms, X-linked dominant hypophosphatemic rickets (most common type), autosomal dominant hypophosphatemic rickets, and autosomal recessive hypophosphatemic rickets.
- FGF-23-independent hypophosphatemic rickets: This type of rickets is not phosphatonin-dependent. Yet, FGF-23 levels are fairly normal. It is characterized by a pathology in the renal tubules that causes phosphate transport defects.
The main causes of rickets worldwide in older infants and toddlers are due to vitamin D deficiency, either due to nutritional deficiency or due to insufficient sun exposure. A study revealed 89% of the patients with rickets had no or minimal sun exposure.[11] Risk factors for nutritional vitamin D deficiency include prolonged exclusive breastfeeding without vitamin D supplementation, excessive juice rather than fortified milk consumption, and inadequate intake of vitamin D fortified foods.[12] Pregnant mothers who have a vitamin D deficiency may predispose their babies to rickets and hypocalcemia.[2][13] Latitude also plays an important role in vitamin D deficiency and rickets. In higher latitudes, there are more chances to develop vitamin D deficiency and rickets.[14] This happens because latitude affects the zenith angle of the sun, and subsequently, the amount of the ultraviolet B (UVB) radiation. Even in the countries where the sunlight is available throughout the year, limited sun exposure can occur due to the following reasons - clothing that covers most of the skin (due to religious, or cultural, or climatic reasons), being indoors for most of the time, having dark skin color (high concentrations of melanin in the skin reduce vitamin D synthesis) or reduced dietary intake (vegetarians)[15], living in a region with high atmospheric pollution (pollutions limit the UVB rays from reaching the ground levels),[16] or extensive use of sunscreens with protection factor > 8.[17] The safe threshold for UVB exposure for vitamin D synthesis without an increased risk for skin cancer is unknown.[18]
Epidemiology
The prevalence of the disease has increased in both developed and developing countries.[19] Yet, generally speaking, the prevalence of rickets is higher in developing counties than in developed countries. African, Middle Eastern, and Asian countries have a wide prevalence rate of 10% to 70%.[6] Historically, the prevalence of rickets in the developed countries significantly decreased following the widespread introduction of dietary vitamin D supplementation, legislation to improve air quality, and recognition of the importance of vitamin D and bone health by the public.[6] In the U.S., vitamin D milk fortification (100 IU/cup) was started in the 1930s.
The worldwide incidence has been estimated differently per 100,000 in various countries as follows: in Canada, 2.9; in New Zealand, 10.5 among children less than three years while it is 2.2 in the older age group (3-15 years); in Australia, 4.9; in Turkey, 3.8; and in the United Kingdom, 7.5.[20][21][22][23][19] In the U.S., specifically in Minnesota, there is a substantial increase in the incidence in the last 20 years. The reported incidences in the 1970s, 1980s, 1990s, and 2000s were 0, 2.2, 3.7, and 24.1, respectively.[24] Restricted sunlight (UVB) exposure is one of the reasons for this increasing prevalence.[6] Native Alaskan children are at considerable risk for developing rickets than other U.S. populations.[14] The incidence of the rickets hospitalization rate is the highest in Alaska among all U.S. regions, 2.23/100,000 versus 1.23/ 100,000.[14] The incidence of rickets in Alaska is significantly high because of higher latitudes.
Pathophysiology
The osseous tissue in the growing long bones is created from the cartilage by a process called endochondral ossification. The chondrocytes in the cartilage grow to form the hypertrophic chondrocytes, which then start producing the cartilage matrix. This cartilage matrix is then calcified, which is reabsorbed and replaced with woven bone, which is later replaced by mature lamellar bone.[2] In these processes, there is a formation of unmineralized bone tissue (osteoid), and the osteoid is mineralized in the presence of adequate calcium and phosphate levels. Any defect in osteoid mineralization may cause rickets.[2] In all types of rickets, the characteristic features occur at the growth plate.
Calcium and phosphorus are required for the normal matrix mineralization. Reduction in these minerals causes abnormal mineralization.[25] Normal serum calcium levels require sufficient dietary calcium intake, normal calcium absorption through the gastrointestinal tract, and adequate active form of vitamin D.
Vitamin D levels are maintained either by endogenous synthesis or dietary intake.
- Endogenous vitamin D synthesis: Vitamin D is synthesized in the human skin after exposure to UVB irradiation (radiation with wavelength 290 to 320 nm).[18]
- Exogenous vitamin sources: Vitamin D can be obtained from dietary sources such as fish olive oil, egg yolk, and fatty fish. However, regular dietary sources may not be adequate to meet the daily needs of vitamin D without extra supplementation or fortification.[4] Some of the foods fortified with vitamin D in the U.S. include milk, margarine, and cereals. Vitamin D supplements are available in two forms, vitamin D2 (ergocalciferol) and vitamin D3 (cholecalciferol).
Low serum calcium, either from its low intake or from vitamin D deficiency, causes a compensatory increase in the PTH, which subsequently causes hypophosphatemia. Low serum phosphate, in turn, inhibits the apoptosis of chondrocytes and thereby accumulation of hypertrophic chondrocytes. Eventually, abnormal growth of the cartilaginous epiphyseal plate occurs. This results in many of the clinical manifestations as well as the radiological changes (widening of epiphysis) of rickets.[8][25] Growth plate abnormalities occur due to reduced vascular invasion with decreased chondroblast and osteoclast activity.
History and Physical
A detailed history and a thorough physical examination are essential to diagnose patients with rickets. History should include the gestational age of the child, details of sunlight exposure, dietary history including intake of supplements, developmental/growth history, and pertinent family history. Positive family history of skeletal abnormalities, stunted growth, alopecia, dental abnormalities, parental consanguinity may suggest a genetic cause of rickets. Physical examinations should include detailed skeletal examination (with attention to any tenderness, deformities, softening, asymmetry, and neurological abnormalities) as well as a detailed dental evaluation.[26] History and physical examination usually give clues to diagnose rickets. However, the absence of clinical signs of rickets doesn’t exclude this diagnosis, especially in the early stages.[5]
The clinical manifestations of rickets are variable based on the underlying etiology, severity, and duration of the disease.[8] Rickets is frequently noted in children between 6 months to 2 years of life. Children frequently have some osseous clinical manifestations (often noted at the sites of rapid bone proliferation):[18]
- Skull: Craniotabes, which is softening of skull bone, seen in infants older than three months of age.[17] Frontal bossing, and wide fontanels are noted.[2][18]
- Chest: Ricketic rosary due to the widening of the costochondral junction, pigeon chest, and Harrison’s groove (a depression at the lower side of the ribcage that occurs as the diaphragm pulls the soft ribcage at its insertion site).[8][18]
- Extremities: The child, mainly during infancy, may present with deformities of the weight-bearing limbs, that mostly involve the rapidly growing bones. The crawling infants may present with upper limb deformities. However, when the child starts walking, the deformities are noticed in the lower limbs.[8][17] The potential lower limb deformities include bow legs (genu varus), knock knees (genu valgus), and joints (knees and ankles) swelling, whereas the deformities of the upper limbs include the wrist widening. The ulna grows relatively rapidly and hence it is significantly affected.[2]
- Spine: Spinal column deformity and kyphosis.
- Other musculoskeletal findings: Gait disturbance, growth retardation, bone softening, bone pain (present as irritability), bone tenderness, a contracted pelvis that may obstruct the labor during adulthood, hypotonia, proximal myopathy, and limb fractures.[4][17][18]
Other manifestations of rickets are as follows:
Evaluation
If the rickets is clinically suspected, biochemical tests and radiological images are the next steps to confirm the diagnosis.
The most important laboratory marker to diagnose the rickets is serum alkaline phosphatase (ALP), which is typically high as this is a disease of abnormal mineralization and increased osteoblastic activity.[5] Alkaline phosphatase activity is induced by phosphate deficiency in rickets.[29] In phosphopenic rickets, ALP values are frequently noted between 400-800 IU/L, and in calcipenic rickets, ALP is markedly elevated, and values are frequently noted up to/greater than 2000 IU/L.[29] It is also an excellent marker to monitor disease activity.
Serum 25-hydroxyvitamin D level is another laboratory marker that helps to diagnose rickets, especially the nutritional deficiency of vitamin D. The active form of vitamin D (1,25-dihydroxy vitamin D) has a short half-life (5-10 hours).[30] Serum 25-hydroxyvitamin D level is the major circulatory form and is typically used to assess vitamin D status.[31] The majority of children with vitamin D deficiency-induced nutritional rickets have serum 25 hydroxyvitamin D level < 10 ng/mL.[8] (The global consensus recommendations on the prevention and treatment of nutritional rickets defined vitamin D deficiency as 25-hydroxyvitamin D level < 30 ng/mL, insufficiency as 30 to 50 ng/mL, and an adequate level as >50 ng/mL.[32]
Routine screening with serum 25 hydroxyvitamin D levels is not recommended for healthy children.[18] Serum 1,25 dihydroxy vitamin D levels are helpful in the evaluation of genetic forms of vitamin D dependent rickets. Generally, 1,25-dihydroxy vitamin D levels are low in vitamin D-dependent rickets type I (A and B) and high in vitamin D-dependent rickets type II (A and B).[10]
Serum calcium and phosphate levels could vary depending on the type of rickets - calcipenic or phosphopenic type. Serum calcium and PTH levels are usually normal in phosphopenic type. In calcipenic rickets, serum calcium levels are either low or normal (as a result of compensatory increases in PTH).[2] If the serum albumin is low, serum calcium levels should be corrected accordingly. In calcipenic rickets, the serum phosphate level is initially normal but drops later in the disease due to renal phosphate loss as a result of elevated PTH.[2]
Measuring urine phosphate is helpful in evaluating the renal loss of phosphate in the genetic forms of hypophosphatemic rickets and other conditions such as Fanconi syndrome associated with phosphaturia.[26] Similarly, urine calcium can be used to monitor hypercalciuria (to prevent nephrocalcinosis) during the treatment phase of hypocalcemic rickets.
Other biochemical investigations include blood urea nitrogen (BUN)/creatinine levels to screen for renal status, and liver enzymes to screen liver function.[26]
The radiological images should include the distal ends of rapidly growing bones in upper and lower extremities[8], and additionally, ribcage images are helpful as well. The appearance of radiolucent lines at the conjunction between epiphysis and metaphysis and widening of the epiphyseal plate, due to the accumulation of non-mineralized osteoid, is the earliest radiological change.[4] Rachitic changes also include the cupping, splaying, fraying, and trabecular formation of the metaphysis.[18] The epiphyseal center formation may be delayed or appear small.[26] The cortex of the bones may be thin and osteopenic.[26][18] Chest images show rachitic rosary and widening of costochondral junctions. Angular deformities, along with pathological fractures of the upper and lower limb bones, may be noted in advanced stages.[26][18]
The combination of positive clinical signs, relevant laboratory findings (high ALP, and either hypocalcemia or hypophosphatemia), and typical radiological findings confirm the diagnosis of rickets. The diagnosis is still possible in the presence of normal serum calcium and phosphate levels. Similarly, clinical signs are not recognized in the early stages.[5]
Treatment / Management
Treatment strategies of rickets depend on the underlying etiology - nutritional vs. genetic rickets.
Treatment of rickets due to nutritional deficiency of vitamin D:
Treatment includes early intensive and late maintenance phase. There are several regimens utilized to treat rickets due to nutritional deficiency of vitamin D.[33] All of them comprise some form of vitamin D administration, vitamin D2 (ergocalciferol) or vitamin D3 (cholecalciferol), with subsequent monitoring for healing.[8] The intensive phase of vitamin D treatment is given for two to three months in conjunction with calcium supplementation (500 mg either through diet or by supplements) for children who have insufficient dietary calcium.[8][18](A1)
- Single-dose therapy (stoss therapy): Here, a single large dose of vitamin D is used, particularly in patients with low medication compliance.[34] It is given orally or intramuscularly at a dose of 100,000 to 600,000 international units (IU) for infants > 1 month.[17] Oral treatment, however, is recommended as this route restores vitamin D more rapidly.[18] This regimen is generally both safe and effective in treating vitamin D deficiency rickets.[34] However, rarely it may be complicated by the increase in the risk of hypercalcemia.[8] The U.S. Endocrine Society recommends a dose of 50,000 IU of vitamin D (either vitamin D2 or D3) once weekly for six weeks, followed by a maintenance phase with 400-600 IU of vitamin D daily. (B3)
- Multiple doses of vitamin D: Small daily doses of vitamin D are given in this regimen. The doses depend on the age of the patient. The daily dose for infants < 1 month, 1-12 months, > 12 months are 1000 IU, 1000 – 5000 IU, and 5000 IU, respectively, for 2-3 months.[17] Subsequently, it is recommended to give a daily dose of 400 IU as the maintenance phase.[17] Almost on similar lines, the U.S. Endocrine Society recommends 2000 IU of vitamin D daily for six weeks in the intensive phase for ages in the intensive therapy and 400-600 IU for the maintenance for vitamin D deficiency.[35] (A1)
In toddlers, bone pain improves within two weeks after treatment initiation.[8] The metaphyseal swelling improves by six months, and the bow legs and knock knee improvement can take up to 2 years. However, adolescents are usually left with some residuals that may need orthopedic surgical correction.[8]
Biochemically, serum calcium, and phosphate levels return to normal levels within six to ten days, whereas PTH normalizes in one to two months. ALP may normalize in three months or longer.[17]
Once the treatment is started, careful monitoring of serum calcium, phosphate, ALP, and 25 hydroxyvitamin D is carried out.[26] Random urine calcium to creatinine ratio should be measured to evaluate any increase in urinary calcium, which is often used to monitor the need to adjust the treatment and also to evaluate for hypercalciuria to prevent nephrocalcinosis. After treatment of the biochemical abnormalities and vitamin D restoration, severe persistent osseous abnormalities may be treated surgically.[26](B3)
Prevention of rickets due to nutritional deficiency of vitamin D: This is a preventable disease. The optimal way to prevent nutritional rickets is to educate the parents and pregnant women about the good dietary sources of calcium and vitamin D as well as about the importance of adequate sun exposure. However, the safe threshold limit for sunlight exposure for vitamin D synthesis without an increased risk for skin cancer is not known.[18] The pregnant women ideally should receive 600 IU per day of vitamin D in combination with other micronutrients to prevent rickets in their offsprings.[18] Vitamin D supplementation during pregnancy helps to avoid high levels of placental ALP and subsequent increase in the neonatal fontanel size, hypocalcemia, and dental enamel complications.[18] (B3)
Additionally, rickets can be prevented by a universal oral vitamin D supplementation of 400 IU given daily to breastfed infants and infants who consume less than 500 mL of fortified formula per day in the first year of their life.[4] Beyond infancy, the high-risk groups for vitamin D deficiency (children with a previous history of rickets, and high risk of insufficient dietary vitamin D) should receive 600 IU of vitamin D daily either by diet or by supplementation.[18] (B3)
Vitamin D supplementation is an effective intervention in preventing vitamin D deficiency rickets. For example, a nationwide program in turkey was introduced in 2005 to propose a free vitamin D drops (400 IU/day) to children less than three years. The prevalence of rickets has dropped from 6% in 1998 to 0.1% in 2008.[36] The residents of the geographical areas located higher than 55th attitudes in Canada were recommended taking higher daily maintenance doses of 800 IU through dietary sources and/or vitamin D supplementation.[14] (B2)
Treatments of rickets due to genetic causes:
- The genetic conditions resulting in rickets are best treated by a pediatric endocrinologist and/or metabolic bone specialist.
- Both vitamin D-dependent rickets type I A (VDDR1A) and vitamin D-dependent rickets type I B (VDDR1B) are treated with calcitriol (1,25-dihydroxyvitamin D).[10]
- Vitamin D-dependent rickets type II A (VDDR2A) and vitamin D-dependent rickets type II B (VDDR2B) are treated with high doses of calcitriol and calcium. The long term management includes a high dose of intravenous calcium administration.[10]
- Familial hypophosphatemic rickets is treated with oral phosphate supplementation along with vitamin D as calcitriol or alfacalcidol (1α-hydroxycholecalciferol).[26] (B3)
Differential Diagnosis
Rickets should be differentiated from the following conditions that mimic based on either the biochemical abnormalities or radiological features:
- Renal insufficiency: Bone disease, also known as renal osteodystrophy, occurs due to many reasons such as the reduced formation of 1,25- dihydroxy vitamin D, metabolic acidosis, retention of phosphate, aluminum toxicity, and secondary hyperparathyroidism.
- Conditions increase ALP: Transient hyperphosphatasemia (a benign condition characterized by an increase in ALP mostly seen in children less than < 5 years of age, where both bone and liver fractions are elevated, however, liver enzymes and 25 hydroxyvitamin D levels will be normal [37]), and cholestatic liver diseases (elevated gamma-glutamyl transferase)
- Conditions which cause hypocalcemia: Primary hypoparathyroidism
- Other bone diseases such: Osteogenesis imperfecta, congenital syphilis
- Nonaccidental injury
- Normal variation: Bowlegs may be seen normally in children < 2 years.[17]
- Skeletal dysplasia: Achondroplasia
- Blount disease: Pathological cox vara (bow legs) due to disruption and altered enchondral bone formation of the medial aspect of proximal metaphysis of the tibia
Prognosis
The prognosis depends on the cause and severity of rickets. Nutritional rickets has a promising prognosis with prompt recognition and early institution of treatment. It can be cured completely within a few months from starting treatment. However, untreated patients may end with catastrophic complications. On the other hand, genetic causes of rickets mostly are not curable, and the treatment is symptomatic to improve the quality of life and management of complications.
Complications
The potential complication of the untreated condition includes poor linear growth, osseous deformities, multiple pathological fractures, hydrocephalus, increased intracranial hypertension (ICH), abnormal dentition (dental caries, dental hypoplasia, delayed dentition). Persistent hypocalcemia can lead to complications such as skeletal and cardiac myopathy, seizures, and eventual death.
Deterrence and Patient Education
The parents should be educated about good dietary sources for vitamin D and calcium, fortified food consumption, and also about adequate sun exposure. Vitamin D supplementation to pregnant women and infants is crucial to prevent this condition. Additionally, this condition would get a great benefit from governmental nutritional assistant programs such as the Supplemental Nutrition Assistance Program (SNAP) in addressing nutritional deficiencies.
Enhancing Healthcare Team Outcomes
Ideally, rickets is treated with a multidisciplinary approach. The treated team ideally should consist of a primary care physician (pediatrician), registered dietitian, and pediatric endocrinologist. Radiologists help in the interpretation of rachitic radiographic features from other conditions. Referral to an orthopedic surgeon may be needed to treat the deformities. Additionally, in genetic cases of rickets, referral to a geneticist in providing genetic counseling for the family. Consultation with a pediatric nephrologist and/or metabolic bone specialist is helpful in genetic conditions. The role of pharmacists is indispensable in helping clinicians in administering medications.[26]) Other specialists, such as cardiologists, neurologists, physical therapists, and occupational therapists, help in the management of associated complications.
Media
(Click Image to Enlarge)
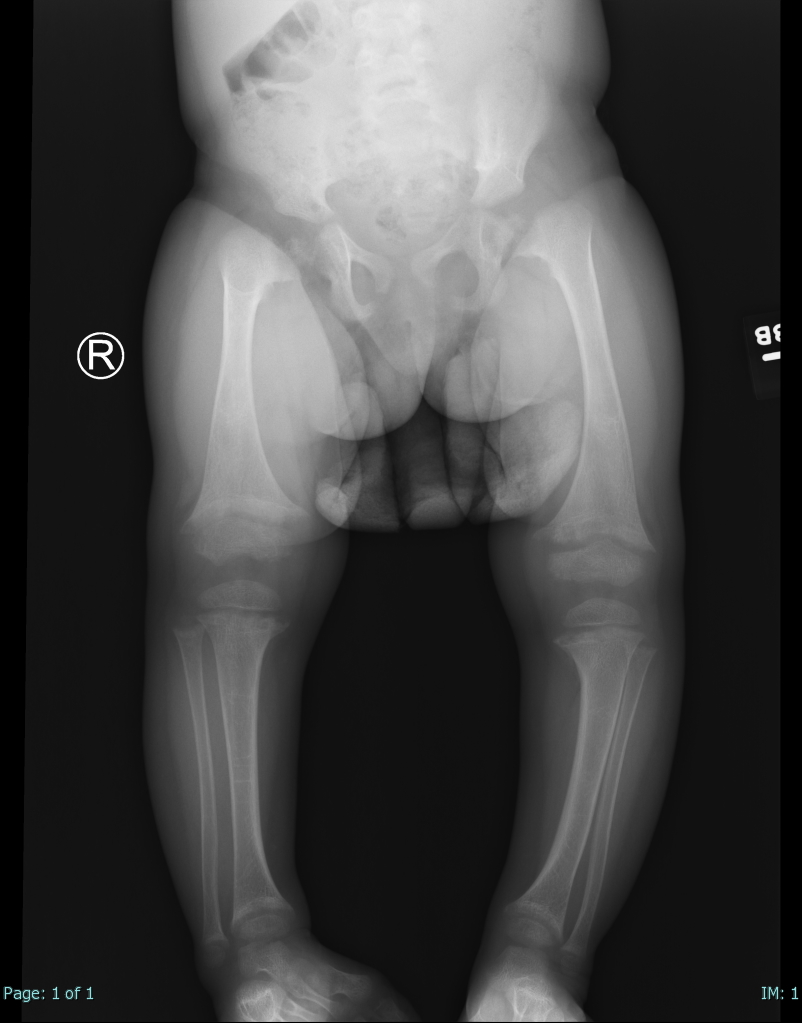
Rickets. Single AP view from a leg alignment study demonstrates metaphyseal flaring (widening) of the distal femurs and proximal and distal tibial metaphyses. Tibial-shaft inbowing is also evident in the X-ray of this 2-year-old female with a known history of rickets.
Contributed by Hassana Barazi, MD
(Click Image to Enlarge)
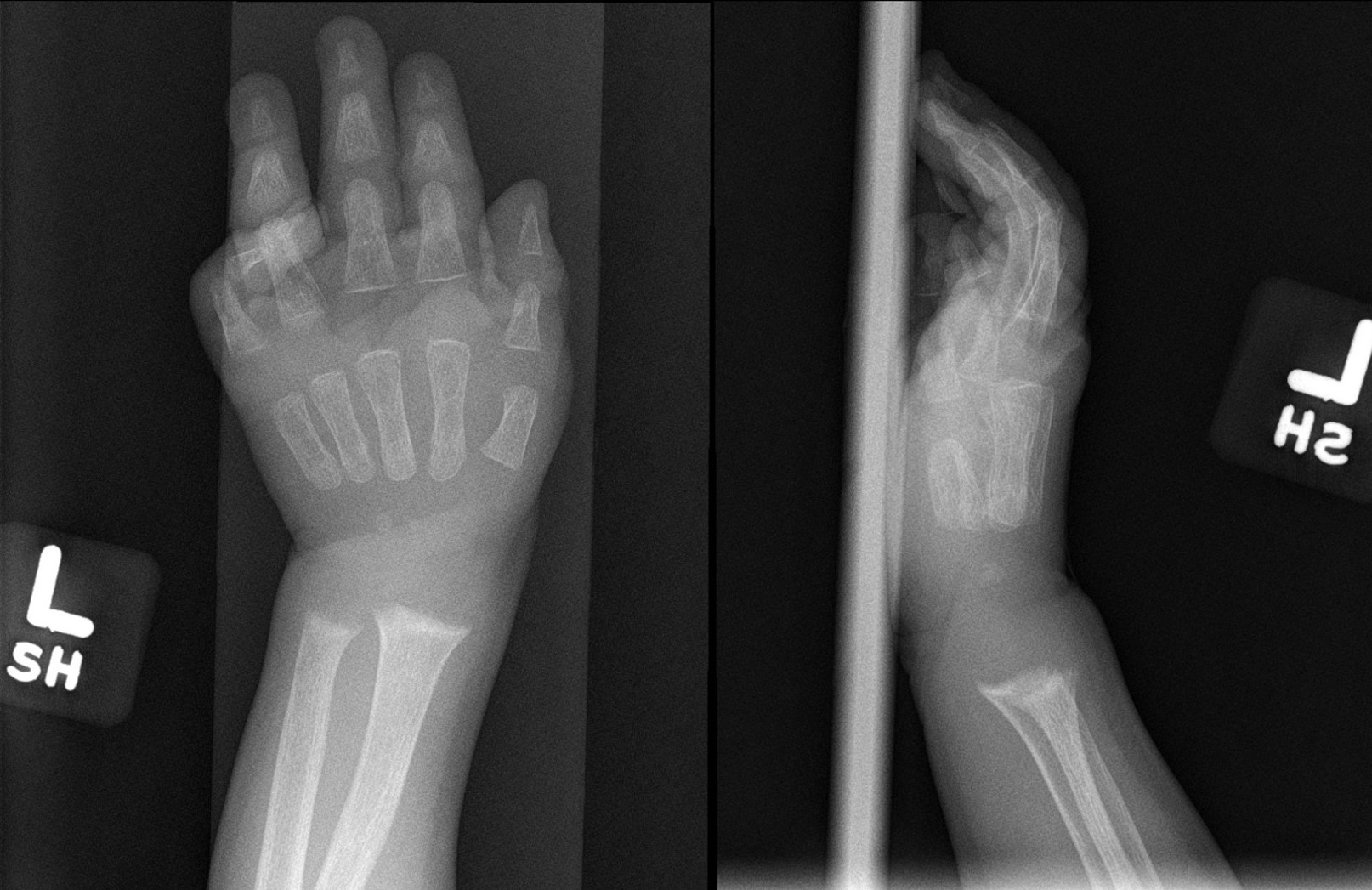
Radiograph of the left wrist (anteroposterior and lateral views) showing metaphyseal widening and irregularity of the distal radius and ulna indicative of rickets Contributed by Senthilkumar Sankararaman, MD
(Click Image to Enlarge)
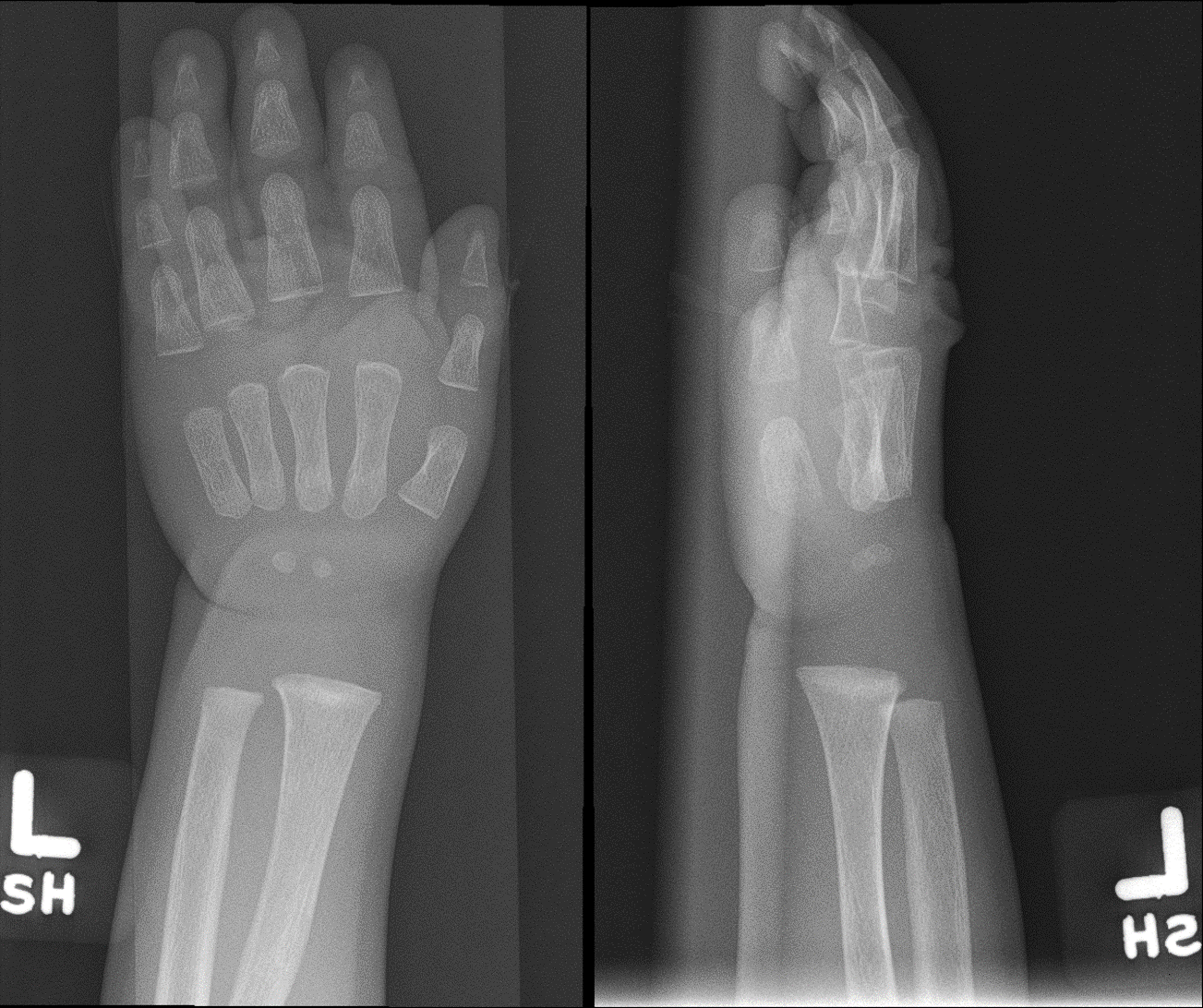
Radiograph of the left wrist (anteroposterior and lateral views) taken three months later showing improved density and appearance of the distal metaphysis after initiation of treatment for rickets. Improved mineralization of the bones also seen. Contributed by Senthilkumar Sankararaman, MD
(Click Image to Enlarge)
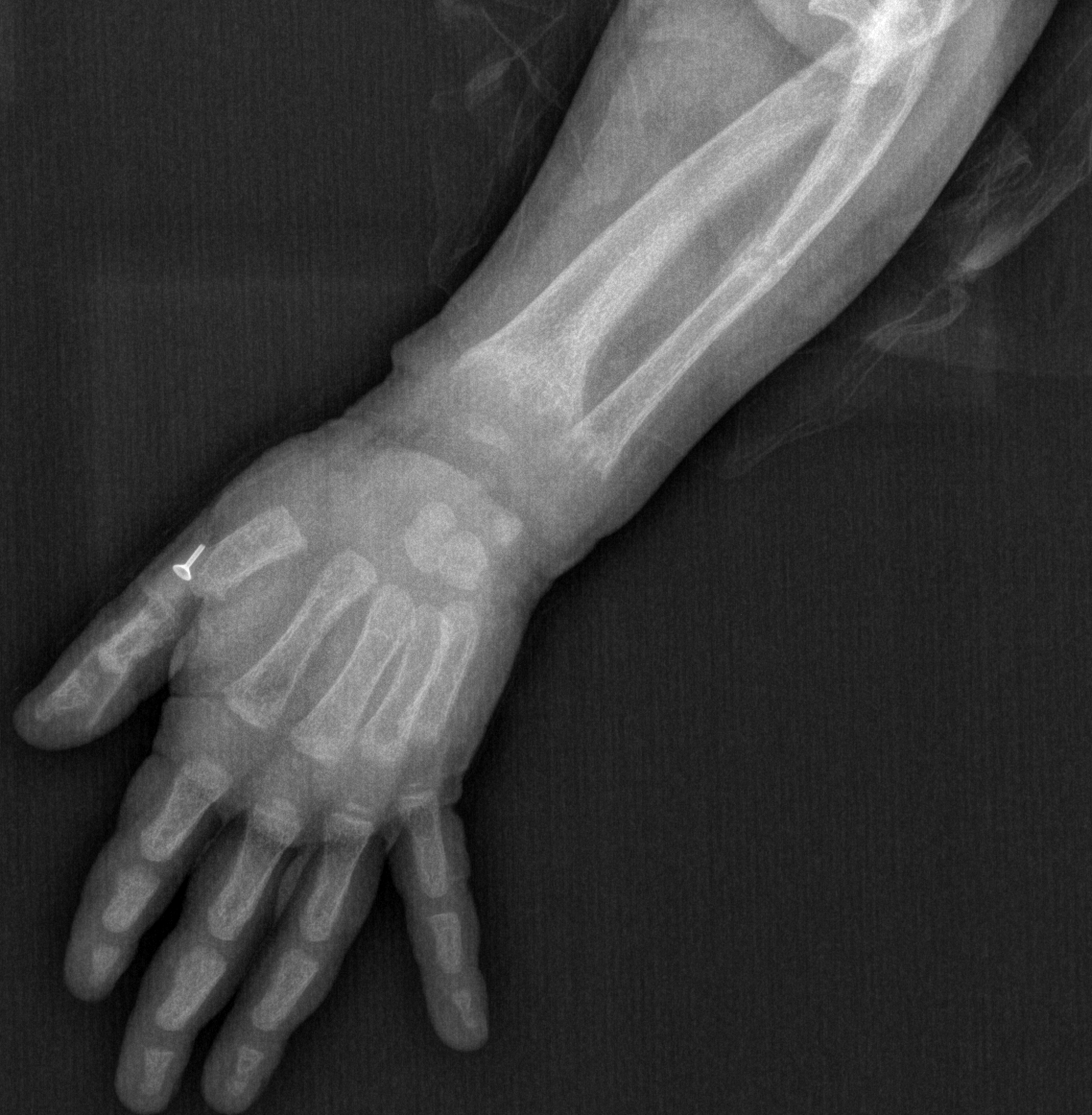
Rickets in a five year old Contributed by Ismail Dursun, MD
References
Pitt MJ. Rickets and osteomalacia are still around. Radiologic clinics of North America. 1991 Jan:29(1):97-118 [PubMed PMID: 1985332]
Level 3 (low-level) evidenceWharton B, Bishop N. Rickets. Lancet (London, England). 2003 Oct 25:362(9393):1389-400 [PubMed PMID: 14585642]
Shaw NJ. Prevention and treatment of nutritional rickets. The Journal of steroid biochemistry and molecular biology. 2016 Nov:164():145-147. doi: 10.1016/j.jsbmb.2015.10.014. Epub 2015 Oct 19 [PubMed PMID: 26493853]
Wheeler BJ, Snoddy AME, Munns C, Simm P, Siafarikas A, Jefferies C. A Brief History of Nutritional Rickets. Frontiers in endocrinology. 2019:10():795. doi: 10.3389/fendo.2019.00795. Epub 2019 Nov 14 [PubMed PMID: 31798536]
Fukumoto S, Ozono K, Michigami T, Minagawa M, Okazaki R, Sugimoto T, Takeuchi Y, Matsumoto T. Pathogenesis and diagnostic criteria for rickets and osteomalacia - proposal by an expert panel supported by Ministry of Health, Labour and Welfare, Japan, The Japanese Society for Bone and Mineral Research and The Japan Endocrine Society. Endocrine journal. 2015:62(8):665-71. doi: 10.1507/endocrj.EJ15-0289. Epub 2015 Jul 4 [PubMed PMID: 26156530]
Prentice A. Nutritional rickets around the world. The Journal of steroid biochemistry and molecular biology. 2013 Jul:136():201-6. doi: 10.1016/j.jsbmb.2012.11.018. Epub 2012 Dec 7 [PubMed PMID: 23220549]
Al-Sharafi BA, Al-Imad SA, Shamshair AM, Al-Faqeeh DH. Severe rickets in a young girl caused by celiac disease: the tragedy of delayed diagnosis: a case report. BMC research notes. 2014 Oct 8:7():701. doi: 10.1186/1756-0500-7-701. Epub 2014 Oct 8 [PubMed PMID: 25292220]
Level 3 (low-level) evidenceMughal MZ. Rickets. Current osteoporosis reports. 2011 Dec:9(4):291-9. doi: 10.1007/s11914-011-0081-0. Epub [PubMed PMID: 21968816]
Michałus I, Rusińska A. Rare, genetically conditioned forms of rickets: Differential diagnosis and advances in diagnostics and treatment. Clinical genetics. 2018 Jul:94(1):103-114. doi: 10.1111/cge.13229. Epub 2018 Mar 25 [PubMed PMID: 29417983]
Level 3 (low-level) evidenceAcar S, Demir K, Shi Y. Genetic Causes of Rickets. Journal of clinical research in pediatric endocrinology. 2017 Dec 30:9(Suppl 2):88-105. doi: 10.4274/jcrpe.2017.S008. Epub 2017 Dec 27 [PubMed PMID: 29280738]
Robinson PD, Högler W, Craig ME, Verge CF, Walker JL, Piper AC, Woodhead HJ, Cowell CT, Ambler GR. The re-emerging burden of rickets: a decade of experience from Sydney. Archives of disease in childhood. 2006 Jul:91(7):564-8 [PubMed PMID: 15956045]
Level 2 (mid-level) evidencePerez-Rossello JM, Feldman HA, Kleinman PK, Connolly SA, Fair RA, Myers RM, Gordon CM. Rachitic changes, demineralization, and fracture risk in healthy infants and toddlers with vitamin D deficiency. Radiology. 2012 Jan:262(1):234-41. doi: 10.1148/radiol.11110358. Epub 2011 Nov 21 [PubMed PMID: 22106354]
Dijkstra SH, van Beek A, Janssen JW, de Vleeschouwer LH, Huysman WA, van den Akker EL. High prevalence of vitamin D deficiency in newborn infants of high-risk mothers. Archives of disease in childhood. 2007 Sep:92(9):750-3 [PubMed PMID: 17715438]
Level 2 (mid-level) evidenceSingleton R, Lescher R, Gessner BD, Benson M, Bulkow L, Rosenfeld J, Thomas T, Holman RC, Haberling D, Bruce M, Bartholomew M, Tiesinga J. Rickets and vitamin D deficiency in Alaska native children. Journal of pediatric endocrinology & metabolism : JPEM. 2015 Jul:28(7-8):815-23. doi: 10.1515/jpem-2014-0446. Epub [PubMed PMID: 25741788]
Level 2 (mid-level) evidenceClemens TL, Adams JS, Henderson SL, Holick MF. Increased skin pigment reduces the capacity of skin to synthesise vitamin D3. Lancet (London, England). 1982 Jan 9:1(8263):74-6 [PubMed PMID: 6119494]
Agarwal KS, Mughal MZ, Upadhyay P, Berry JL, Mawer EB, Puliyel JM. The impact of atmospheric pollution on vitamin D status of infants and toddlers in Delhi, India. Archives of disease in childhood. 2002 Aug:87(2):111-3 [PubMed PMID: 12138058]
Level 2 (mid-level) evidenceOzkan B. Nutritional rickets. Journal of clinical research in pediatric endocrinology. 2010:2(4):137-43. doi: 10.4274/jcrpe.v2i4.137. Epub 2010 Nov 1 [PubMed PMID: 21274312]
Munns CF, Shaw N, Kiely M, Specker BL, Thacher TD, Ozono K, Michigami T, Tiosano D, Mughal MZ, Mäkitie O, Ramos-Abad L, Ward L, DiMeglio LA, Atapattu N, Cassinelli H, Braegger C, Pettifor JM, Seth A, Idris HW, Bhatia V, Fu J, Goldberg G, Sävendahl L, Khadgawat R, Pludowski P, Maddock J, Hyppönen E, Oduwole A, Frew E, Aguiar M, Tulchinsky T, Butler G, Högler W. Global Consensus Recommendations on Prevention and Management of Nutritional Rickets. Hormone research in paediatrics. 2016:85(2):83-106. doi: 10.1159/000443136. Epub 2016 Jan 8 [PubMed PMID: 26741135]
Level 3 (low-level) evidenceMunns CF, Shaw N, Kiely M, Specker BL, Thacher TD, Ozono K, Michigami T, Tiosano D, Mughal MZ, Mäkitie O, Ramos-Abad L, Ward L, DiMeglio LA, Atapattu N, Cassinelli H, Braegger C, Pettifor JM, Seth A, Idris HW, Bhatia V, Fu J, Goldberg G, Sävendahl L, Khadgawat R, Pludowski P, Maddock J, Hyppönen E, Oduwole A, Frew E, Aguiar M, Tulchinsky T, Butler G, Högler W. Global Consensus Recommendations on Prevention and Management of Nutritional Rickets. The Journal of clinical endocrinology and metabolism. 2016 Feb:101(2):394-415. doi: 10.1210/jc.2015-2175. Epub 2016 Jan 8 [PubMed PMID: 26745253]
Level 3 (low-level) evidenceWard LM, Gaboury I, Ladhani M, Zlotkin S. Vitamin D-deficiency rickets among children in Canada. CMAJ : Canadian Medical Association journal = journal de l'Association medicale canadienne. 2007 Jul 17:177(2):161-6 [PubMed PMID: 17600035]
Wheeler BJ, Dickson NP, Houghton LA, Ward LM, Taylor BJ. Incidence and characteristics of vitamin D deficiency rickets in New Zealand children: a New Zealand Paediatric Surveillance Unit study. Australian and New Zealand journal of public health. 2015 Aug:39(4):380-3. doi: 10.1111/1753-6405.12390. Epub 2015 Jun 29 [PubMed PMID: 26122859]
Munns CF, Simm PJ, Rodda CP, Garnett SP, Zacharin MR, Ward LM, Geddes J, Cherian S, Zurynski Y, Cowell CT, APSU Vitamin D Study Group. Incidence of vitamin D deficiency rickets among Australian children: an Australian Paediatric Surveillance Unit study. The Medical journal of Australia. 2012 Apr 16:196(7):466-8 [PubMed PMID: 22509879]
Callaghan AL, Moy RJ, Booth IW, Debelle G, Shaw NJ. Incidence of symptomatic vitamin D deficiency. Archives of disease in childhood. 2006 Jul:91(7):606-7 [PubMed PMID: 16595644]
Thacher TD, Fischer PR, Tebben PJ, Singh RJ, Cha SS, Maxson JA, Yawn BP. Increasing incidence of nutritional rickets: a population-based study in Olmsted County, Minnesota. Mayo Clinic proceedings. 2013 Feb:88(2):176-83. doi: 10.1016/j.mayocp.2012.10.018. Epub [PubMed PMID: 23374621]
Goltzman D. Functions of vitamin D in bone. Histochemistry and cell biology. 2018 Apr:149(4):305-312. doi: 10.1007/s00418-018-1648-y. Epub 2018 Feb 12 [PubMed PMID: 29435763]
Nield LS, Mahajan P, Joshi A, Kamat D. Rickets: not a disease of the past. American family physician. 2006 Aug 15:74(4):619-26 [PubMed PMID: 16939184]
Level 3 (low-level) evidenceHanafy MM, Hassanein ES, el Khateeb S. Benign intracranial hypertension in vitamin D deficiency rickets associated with malnutrition. The Journal of tropical pediatrics and African child health. 1967 Mar:13(1):19-22 [PubMed PMID: 5298825]
Vakharia JD, Matlock K, Taylor HO, Backeljauw PF, Topor LS. Craniosynostosis as the Presenting Feature of X-linked Hypophosphatemic Rickets. Pediatrics. 2018 Apr:141(Suppl 5):S515-S519. doi: 10.1542/peds.2017-2522. Epub [PubMed PMID: 29610183]
Lambert AS, Linglart A. Hypocalcaemic and hypophosphatemic rickets. Best practice & research. Clinical endocrinology & metabolism. 2018 Aug:32(4):455-476. doi: 10.1016/j.beem.2018.05.009. Epub 2018 Jul 4 [PubMed PMID: 30086869]
Bailie GR, Johnson CA. Comparative review of the pharmacokinetics of vitamin D analogues. Seminars in dialysis. 2002 Sep-Oct:15(5):352-7 [PubMed PMID: 12358640]
Level 2 (mid-level) evidenceGreer FR. Vitamin D deficiency--it's more than rickets. The Journal of pediatrics. 2003 Oct:143(4):422-3 [PubMed PMID: 14571210]
Chanchlani R, Nemer P, Sinha R, Nemer L, Krishnappa V, Sochett E, Safadi F, Raina R. An Overview of Rickets in Children. Kidney international reports. 2020 Jul:5(7):980-990. doi: 10.1016/j.ekir.2020.03.025. Epub 2020 Apr 11 [PubMed PMID: 32647755]
Level 3 (low-level) evidenceGordon CM, Williams AL, Feldman HA, May J, Sinclair L, Vasquez A, Cox JE. Treatment of hypovitaminosis D in infants and toddlers. The Journal of clinical endocrinology and metabolism. 2008 Jul:93(7):2716-21. doi: 10.1210/jc.2007-2790. Epub 2008 Apr 15 [PubMed PMID: 18413426]
Level 1 (high-level) evidenceShah BR, Finberg L. Single-day therapy for nutritional vitamin D-deficiency rickets: a preferred method. The Journal of pediatrics. 1994 Sep:125(3):487-90 [PubMed PMID: 8071764]
Holick MF, Binkley NC, Bischoff-Ferrari HA, Gordon CM, Hanley DA, Heaney RP, Murad MH, Weaver CM, Endocrine Society. Evaluation, treatment, and prevention of vitamin D deficiency: an Endocrine Society clinical practice guideline. The Journal of clinical endocrinology and metabolism. 2011 Jul:96(7):1911-30. doi: 10.1210/jc.2011-0385. Epub 2011 Jun 6 [PubMed PMID: 21646368]
Level 1 (high-level) evidenceHatun Ş, Ozkan B, Bereket A. Vitamin D deficiency and prevention: Turkish experience. Acta paediatrica (Oslo, Norway : 1992). 2011 Sep:100(9):1195-9. doi: 10.1111/j.1651-2227.2011.02383.x. Epub 2011 Jul 4 [PubMed PMID: 21672012]
Skalova S, Kutilek S. Transient hyperphosphatemia: a benign laboratory disorder in a boy with Gitelman syndrome. Jornal brasileiro de nefrologia. 2016 Jul-Sep:38(3):363-365. doi: 10.5935/0101-2800.20160055. Epub [PubMed PMID: 27737396]